USGS Pfe , Science for a Changing World \ \C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2025 Metropolitan Transportation Plan
Wilmington Area Planning Council 2025 Update CONNECTIONS to the st 21 Century 2025 Metropolitan Transportation Plan Adopted February 23, 2000 Wilmington Area Planning Council 850 Library Avenue, Suite 100 Newark, DE 19711 Phone (302) 737-6205 Email [email protected] Fax (302) 737-9584 Web www.wilmapco.org Wilmington Area Planning Council * Council: Anne P. Canby, Secretary, Delaware Department of Transportation, Chairperson Robert J. Alt, Mayor, City of Elkton, Vice Chair Nelson K. Bolender, President, Cecil County Commissioners Jeffrey W. Bullock, Chief of Staff, Delaware Governor’s Office Thomas P. Gordon, County Executive, New Castle County James F. Grant, Mayor, Town of Odessa Marsha J. Kaiser, Director, Maryland Department of Transportation Office of Planning & Capital Programming Raymond C. Miller, Director, Delaware Transit Corporation James H. Sills, Jr., Mayor, City of Wilmington Technical Advisory Committee: Public Advisory Committee: Anna Marie Gonnella, Delaware River & Bay Authority, Chairperson Anita Puglisi, City of Newark Citizen, Chairperson Phil Wheeler, Delaware Department of Natural Resources & Barbara Washam, Upper East Side Neighborhood Association, Vice Chair Environmental Control, Vice Chair Doug Andrews, Delmarva Rail Passenger Association Eugene E. Abbott, Delaware Department of Transportation David Blankenship, Wilmington Department of Public Works David Baker, Southern New Castle County Citizen Anthony J. Di Giacomo, Cecil County Office of Planning and Jan Baty, Newark Planning Commission Zoning & Parks and Recreation Lynn Broaddus, Brandywine Hundred Citizen Bobbie Geier, Delaware Transit Corporation Harry Brown, City of Wilmington Citizen Markus R. Gradecak, Maryland Office of Planning John J. Casey, Delaware Contractors Association Herbert M. Inden, Delaware Office of State Planning Bob Dietrich, Delaware Motor Transport Association Coordination Gwinneth Kaminsky Rivera, Wilmington Department of Planning Dennis Flint, White Clay Creek Bicycle Club James P. -

Delaware's 2009 HPMS Submission Comments
May 26, 2010 Office of Highway Policy Information Federal Highway Administration 400 Seventh Street, S.W. Washington, D.C. 20590 Attention: HPPI-20, Room 3306 RE: Delaware HPMS 2009 Dear Sir/Madam: We are submitting herewith the 2009 HPMS Data in accordance with the reporting requirements. Area and Population The 2009 HPMS information reflects the 2000-Census Urban Boundaries. The Land area and Population are presented below: Area (Square Location Miles) 2009 Population Middletown, DE 3.5 6,829 Dover, DE 58.6 73,836 Georgetown, DE 3.6 8,618 Lewes, DE 17.3 18,550 Long Neck, DE 11.8 10,045 Milford, DE - Kent 5.5 5,866 Milford, DE - Sussex 6.3 8,774 Ocean View, DE 10.4 9,965 Philadelphia, PA--NJ--DE--MD 188.2 495,025 Salisbury, MD--DE 0.6 1,276 Seaford, DE 15.6 23,707 Smyrna, DE - Kent 6 16,045 Smyrna, DE - New Castle 1.1 72 Rural 1,625.50 206,692 Total Urban 328.5 678,607 Total Rural 1,625.50 202,925 Total State 1,954 881,532 2009 Delaware HPMS Comments May 26, 2010 Page 2 of 58 The Land Area was calculated in accordance with HPMS guidelines. This year’s calculations match the 2000 Census measurements of Delaware’s total land area of 1,954 square miles. Based on the 2000 Census data, the Delaware Population Consortium provided the following population data and forecasts for each county of the State. Area (Square County Population Miles) 2000 2007 2008 2009 Census New Castle 427 500,265 525,624 528,536 532,083 Kent 589 126,697 151,505 155,142 157,430 Sussex 938 156,638 187,952 188,597 192,019 Total State 1954 783,600 865,081 872,275 881,532 2009 Delaware Certification Public Mileage On May 7, 2010, Delaware reported 2009 Certified Public Miles to the FHWA. -

Elkton Pedestrian Plan
July 2018 2 Elkton Pedestrian Plan Table of Contents Who is WILMAPCO? ..................................................................................................................................... 3 Executive Summary ...................................................................................................................................... 4 Background ................................................................................................................................................... 5 Transportation History of Elkton .............................................................................................................. 5 Purpose of the Study ................................................................................................................................. 6 Previous Studies Completed in Elkton ...................................................................................................... 7 Steering Committee and Timeline .......................................................................................................... 10 Pedestrian Prioritization ............................................................................................................................ 11 Methodology ........................................................................................................................................... 11 Results .................................................................................................................................................... -

Lessons About Protecting the Natural Environment by Controlling Industrial Development Under Delaware's Coastal Zone Act
Pace Environmental Law Review Volume 25 Issue 1 Winter 2008 Article 2 January 2008 Keeping the Coast Clear: Lessons about Protecting the Natural Environment by Controlling Industrial Development under Delaware's Coastal Zone Act Kenneth T. Kristl Follow this and additional works at: https://digitalcommons.pace.edu/pelr Recommended Citation Kenneth T. Kristl, Keeping the Coast Clear: Lessons about Protecting the Natural Environment by Controlling Industrial Development under Delaware's Coastal Zone Act, 25 Pace Envtl. L. Rev. 37 (2008) Available at: https://digitalcommons.pace.edu/pelr/vol25/iss1/2 This Article is brought to you for free and open access by the School of Law at DigitalCommons@Pace. It has been accepted for inclusion in Pace Environmental Law Review by an authorized administrator of DigitalCommons@Pace. For more information, please contact [email protected]. Keeping the Coast Clear: Lessons About Protecting the Natural Environment by Controlling Industrial Development Under Delaware's Coastal Zone Act KENNETH T. KRISTL* In 1970, as the nation celebrated the first Earth Day, the State of Delaware stood at a proverbial fork in the road concerning its then largely undeveloped coastal area. Shell Oil was poised to begin construction of Delaware's second large oil refinery, this one to be located next to the Bombay Hook National Wildlife Refuge.1 Zapata Norness, a bulk shipping company, was planning to build a 300 acre island in the Delaware Bay for staging shipments of bulk quantities of coal and iron ore. 2 The United States Depart- ment of Commerce was working with oil and shipping companies to make the Delaware Bay "the premier supertanker port and in- dustrial center in the East.' 3 Thus, Delaware had a choice: to in- dustrialize its coastal areas (much like its neighbors New Jersey and Pennsylvania had done farther up the Delaware River), or to restrict such industrialization and preserve the natural resources of the coast. -

OCR Document
NPS Form 10-900 (7-81) United States Department of the Interior National Park Service National Register of Historic Places Inventory-Nomination Form See instructions In How to Complete National Register Forms Type all entries--complete applicable sections 1. Name historic _ and/or common Delaware City Historic District 2. Location street & number NA NA not for publication city, town Delaware City vicinity of New Castle 002 state Delaware code 10 county code 3. Classification Category Ownership Status Present Use agriculture X district public X occupied museum X commercial building(s) private X unoccupied park educational structure X both X work In progress X private residence entertainment site Public Acquisition Accessible X religious yes: restricted X government object NA in process scientific being considered X yes: unrestricted Industrial X transportation no military' other: 4. Owner of Property name Multiple (see owners list) street & number state city, town vicinity of 5. Location of Legal Description courthouse, registry of deeds, etc. New Castle County Recorder of Deeds street & number City/County Building, 800 French Street state Delaware city, town Wilmington 6. Representation in Existing Surveys Delaware Cultural Resource title Survey (N-6333) has this property been determined eligible? yes X no date 1979 federal X state county local Bureau of Archeology and Historic Preservation depository for survey records Old State House, The Green city, town Dover state Delaware 7. Description Condition Check one Check one X excellent deteriorated unaltered X original site X good ruins X altered moved date X fair unexposed Describe the present and original (If known) physical appearance The Delaware City Historic District is located within the limits of Delaware City, an incorporated town of approximately 1,800 people that is situated in the eastern central area of New Castle County. -

Fort-Dupont TIS Review Letter
June 9, 2017 Mr. Troy Brestel Project Engineer Development Coordination DelDOT Division of Planning P O Box 778 Dover, DE 19903 RE: Agreement No. 1774 Project Number T201769002 Traffic Impact Study Services Task 4A-Fort DuPont Redevelopment Dear Mr. Brestel: Johnson, Mirmiran and Thompson (JMT) has completed the review of the Traffic Impact Study (TIS) for the Fort DuPont Redevelopment, prepared by Duffield Associates, Inc. This task was assigned Task Number 4A. Duffield Associates, Inc. prepared the report in a manner generally consistent with DelDOT’s Development Coordination Manual. The TIS evaluates the impacts of a mixed-use development proposed in Delaware City, New Castle County, on the east side of Delaware Route 9 (New Castle Road 2) between the Chesapeake and Delaware Canal and the Delaware City Branch Canal. Subsequent to the October 19, 2016, Scoping Meeting at DelDOT, the proposed uses have been modified. Per the TIS, the mixed use consists of 50 single-family detached houses, 105 townhouses, 100 condominiums, 161 low-rise apartment units, an 85-bed assisted care facility, a 25-room hotel, a 13,000 square-foot museum, 141,000 square feet of office space, a 4,720 square-foot government office building, 75,572 square feet of retail, and a 9,800 square-foot quality restaurant. The development will be constructed in two separate phases where Phase 1 will consist of 42 single-family detached houses and 49 townhouses. Phase 1 is expected to be constructed by 2020 and the final phase of construction is anticipated to be completed by 2024. Two access points are proposed for the development: a roundabout at the intersection of Delaware Route 9/New Castle Avenue/Polktown Place (New Castle Road 2A) and a full access at the intersection of Wilmington Avenue/Polktown Place. -

Brittany Danahy Sperry Vana Nessd Vadvisorsisors
LEWES, DELAWARE LEGEND LEGEND PROPERTIES PROPERTIES INC. C O M M E R C I A L R E A L E S T A T E COMMERCIAL REAL ESTATE 8 5 6 2 3 1 1 0 1 0 • w w w . l p r e . c o m the lifestyle center for livable delaware The Vineyards is an 82 acre mixed use development featuring 425,000 sq ft of retail and office space with up to 927 residential units and a 150-room Hilton Garden Inn, plus 35,000 civic space, fitness center, lake, boardwalk and am- phitheater located next to Delaware's oldest and most vibrant vineyard. The Vineyards satisfies the unfulfilled demand for fashion, accessories, home furnishings, dining, and entertainment experiences with the allure of an operational and historic vineyard. This lifestyle center leverages Sussex County’s housing boom which boasts average residential sales of approximately 7,000 units annually with 79,000 housing units proposed or constructed. Huge seasonal population base includes 6,000,000 beach visitors of which 3,000,000 stay overnight. There are 300,000 people within this market, with 140,000 employees. LEGEND LEGEND PROPERTIES PROPERTIES INC. C O M M E R C I A L R E A L E S T A T E COMMERCIAL REAL ESTATE 8 5 6 2 3 1 1 0 1 0 • w w w . l p r e . c o m Sussex County Delaware Demographic Trends Population Growth Rate Per Annum: 3.3% 2000 2007 2012 Leisure Travelers ( > 50 miles) Population 156,638 191,685 219,226 6 million visitors spend average of $328 per visit Households 62,577 77,504 89,265 Quarterly Percentage of Leisure Travel Median HHI $39,138 $50,067 $59,690 Jan-Mar 20% Jul-Sep 35% Source: 2000 Census. -

New Castle Avenue Redevelopment TIS Review Letter
August 25, 2020 Mr. Michael Kaszyski Duffield Associates, Inc. 5400 Limestone Road Wilmington, Delaware 19808 Dear Mr. Kaszyski: The Department has completed its review of the traffic impact study (TIS) for the proposed New Castle Avenue Redevelopment. The TIS was prepared by Duffield Associates, Inc. (DA), and is dated May 2020. DA prepared the report in a manner generally consistent with DelDOT’s Development Coordination Manual. The TIS evaluates the traffic impacts of the proposed development, proposed to be located along the south side of the northbound I-295 off-ramp and northwest of the intersection of New Castle Avenue (Delaware Route 9) and Cherry Lane in New Castle County, Delaware. The proposed development would consist of a 5,200 square foot high-turnover sit-down restaurant, a 2,400 square foot fast-food restaurant with drive-through window, a 2,400 square foot coffee / donut shop with drive-through window, a 4,160 square-foot variety store, and an 88- room business hotel on an approximately 6.07-acre assemblage of parcels (Tax Parcels 1001500001 and 1001500005), and would replace a now-vacant Harley-Davidson dealership. One full access is proposed on New Castle Avenue by way of Cherry Lane. Construction is anticipated to be complete in 2022. The subject land is currently zoned CR (Commercial - Regional) in New Castle County, and the developer does not plan to change the zoning. While DelDOT does not have any current projects in the study area, WILMAPCO has completed a master plan that includes the study area, the Delaware Route 9 Corridor Land Use and Transportation Master Plan (Route 9 Master Plan). -
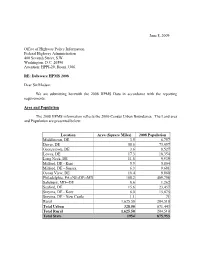
Delaware's 2008 HPMS Submission Comments
June 8, 2009 Office of Highway Policy Information Federal Highway Administration 400 Seventh Street, S.W. Washington, D.C. 20590 Attention: HPPI-20, Room 3306 RE: Delaware HPMS 2008 Dear Sir/Madam: We are submitting herewith the 2008 HPMS Data in accordance with the reporting requirements. Area and Population The 2008 HPMS information reflects the 2000-Census Urban Boundaries. The Land area and Population are presented below: Location Area (Square Miles) 2008 Population Middletown, DE 3.5 6,757 Dover, DE 58.6 73,057 Georgetown, DE 3.6 8,527 Lewes, DE 17.3 18,354 Long Neck, DE 11.8 9,939 Milford, DE - Kent 5.5 5,804 Milford, DE - Sussex 6.3 8,681 Ocean View, DE 10.4 9,860 Philadelphia, PA--NJ--DE--MD 188.2 489,798 Salisbury, MD--DE 0.6 1,262 Seaford, DE 15.6 23,457 Smyrna, DE - Kent 6.0 15,875 Smyrna, DE - New Castle 1.1 71 Rural 1,625.50 204,510 Total Urban 328.50 671,443 Total Rural 1,625.50 204,510 Total State 1954 875,953 2008 Delaware HPMS Comments June 8, 2009 Page 2 of 61 The Land Area was calculated in accordance with HPMS guidelines. This year’s calculations match the 2000 Census measurements of Delaware’s total land area of 1954 square miles. Based on the 2000 Census data, the Delaware Population Consortium provided the following population data and forecasts for each county of the State. Area (Square County Population Miles) 2000 2006 2007 2008 Census New Castle 427 500,265 525,578 529,590 532,057 Kent 589 126,697 147,587 150,516 155,299 Sussex 938 156,638 180,282 183,798 188,597 Total State 1954 783,600 853,447 863,904 875,953 2008 Delaware Certification Public Mileage On May 1, 2009, Delaware reported 2008 Certified Public Miles to the FHWA. -

The Southern New Castle County Scenic River and Highway Study
The Southern New Castle County Scenic River and Highway Study THE SOUTHERN NEW CASTLE COUNTY SCENIC RIVER AND HIGHWAY STUDY New Castle County Department of Land Use 1 The Southern New Castle County Scenic River and Highway Study This publication is the most recent in a series of studies that have been compiled to catalog New Castle County’s scenic and historic resources. Christopher A. Coons, County Executive County Council Paul Clark, President Joseph M. Reda, District 1 George Smiley, District 7 Robert S. Weiner, District 2 John J. Cartier, District 8 William J. Tansey, District 3 Timothy P. Sheldon, District 9 Penrose Hollins, District 4 Jea P. Street, District 10 Stephanie A. McClellan, District 5 David L. Tackett, District 11 William E. Powers, Jr., District 6 James W. Bell, District 12 Charles L. Baker, General Manager, Land Use Department 2 The Southern New Castle County Scenic River and Highway Study The Southern New Castle County Scenic River and Highway Study Prepared by The New Castle County Department of Land Use New Castle County, Delaware In conjunction with Gaadt Perspectives, LLC Chadds Ford, Pennsylvania Historic resource information and analysis provided by Center for Historic and Architectural Design (CHAD) University of Delaware Support provided by Wilmington Metropolitan Area Planning Council (WILMAPCO) New Castle County January 2008 3 The Southern New Castle County Scenic River and Highway Study Introduction and Executive Summary PURPOSE The Southern New Castle County Scenic River and Highway Study follows in the tradition of similar studies executed for the Brandywine and Red Clay Valleys north of the C&D Canal. -

Delaware City Transportation Plan
www.wilmapco.org/DelawareCity DELAWARE CITY TRANSPORTATION PLAN PREPARED BY WILMAPCO FOR DELAWARE CITY Approved April 13, 2009 by Delaware City Council Approved May 14, 2009 by WILMAPCO Council INTRODUCTION .......................................................................................................................................................3 REGIONAL CONTEXT.............................................................................................................................................6 LOCAL CONTEXT – ISSUES AND OPPORTUNITIES .....................................................................................12 ROADWAY CIRCULATION..................................................................................................................................22 PARKING ..................................................................................................................................................................26 PEDESTRIAN AND BICYCLE FACILITIES.......................................................................................................27 TRANSIT ...................................................................................................................................................................30 MOBILITY FRIENDLY DESIGN ..........................................................................................................................32 IMPLEMENTATION ACTION PLAN ..................................................................................................................33 -

Little Creek Hundred Rural Historic District______2
NPS Form 10-900 0MB No. 1024-0018 (3-82) Exp. 10-31-84 United States Department of the Interior National Park Service For NPS use only National Register of Historic Places received SEP 20 1934 Inventory Nomination Form dateentered Nn 7 j4 See instructions in How to Complete National Register Forms Type all entries complete applicable sections____________________________________ 1. Name_____________________________ historic______________"_____________________________________________________ and/or common Little Creek Hundred Rural Historic District_______________________ 2. Location ,,, street & number not for publication city, town Little Creek vicinity of state Delaware code 10 county Kent code 001 3. Classification Category Ownership Status Present Use x district public x occupied x agriculture x museum building(s) private unoccupied commercial park structure x both work in progress educational x private residence site Public Acquisition Accessible entertainment religious object in process yes: restricted government scientific being considered x yes: unrestricted industrial transportation x NA no military other; 4. Owner of Property name Multiple (see attached continuation sheet) street & number city, town vicinity of state 5. Location of Legal Description courthouse, registry of deeds, etc. Kent County Register of Deeds street & number Kent County Building city, town Dover state Delaware 6. Representation in Existing Surveys title Cultural Resource Survey K-5686 has this property been determined eligible? yes x no date 1981 federal x . state county local depository for survey records Bureau of Archaeology and Historic Preservation Dover Delaware city, town state Condition Check Check x excellent *!__ deteriorated ..._x. unaltered x original site ——————x good3? ~" "" __ ruins _-X_ altered moved date _x__ fair __ unexposed original (if The Little Creek Hundred Rural Historic District is located in southern Little Creek Hundred with a small portion in East Dover Hundred, Kent County.