Common and Mutation Specific Phenotypes of KRAS and BRAF
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -
Overexpression of DCLK1-AL Increases Tumor Cell Invasion, Drug Resistance, and KRAS Activation and Can Be Targeted to Inhibit Tumorigenesis in Pancreatic Cancer
Hindawi Journal of Oncology Volume 2019, Article ID 6402925, 11 pages https://doi.org/10.1155/2019/6402925 Research Article Overexpression of DCLK1-AL Increases Tumor Cell Invasion, Drug Resistance, and KRAS Activation and Can Be Targeted to Inhibit Tumorigenesis in Pancreatic Cancer Dongfeng Qu ,1,2,3 Nathaniel Weygant,1 Jiannan Yao ,4 Parthasarathy Chandrakesan,1,2,3 William L. Berry,5 Randal May ,1,2 Kamille Pitts,1 Sanam Husain,6 Stan Lightfoot,6 Min Li,1 Timothy C. Wang,7 Guangyu An ,4 Cynthia Clendenin,8 Ben Z. Stanger,8 and Courtney W. Houchen 1,2,3 Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA Department of Veterans Affairs Medical Center, Oklahoma City, OK, USA Peggy and Charles Stephenson Cancer Center, Oklahoma City, OK, USA Department of Oncology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, China Department of Cell Biology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA Department of Digestive and Liver Diseases, Columbia University Medical Center, New York, NY, USA Department of Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA Correspondence should be addressed to Dongfeng Qu; [email protected] and Courtney W. Houchen; [email protected] Received 24 January 2019; Revised 10 May 2019; Accepted 27 May 2019; Published 5 August 2019 Academic Editor: Francesca De Felice Copyright © 2019 Dongfeng Qu et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -

Brief Genetics Report Haplotype Structures and Large
Brief Genetics Report Haplotype Structures and Large-Scale Association Testing of the 5 AMP-Activated Protein Kinase Genes PRKAA2, PRKAB1, and PRKAB2 With Type 2 Diabetes Maria W. Sun,1,2 Jennifer Y. Lee,1,2 Paul I.W. de Bakker,1,2,3 Noe¨l P. Burtt,2 Peter Almgren,4 Lennart Råstam,5 Tiinamaija Tuomi,6 Daniel Gaudet,7 Mark J. Daly,2,8 Joel N. Hirschhorn,2,3,9 David Altshuler,1,2,3,8,10 Leif Groop,4,6 and Jose C. Florez1,2,8,10 AMP-activated protein kinase (AMPK) is a key molecular plasma glucose, or insulin sensitivity. Several nominal asso- regulator of cellular metabolism, and its activity is induced ciations of variants in PRKAA2 and PRKAB1 with BMI appear by both metformin and thiazolidinedione antidiabetic med- to be consistent with statistical noise. Diabetes 55:849–855, ications. It has therefore been proposed both as a putative 2006 agent in the pathophysiology of type 2 diabetes and as a valid target for therapeutic intervention. Thus, the genes that encode the various AMPK subunits are intriguing ype 2 diabetes arises from the complex interplay candidates for the inherited basis of type 2 diabetes. We therefore set out to test for the association of common of various pathophysiologic mechanisms involv- variants in the genes that encode three selected AMPK ing peripheral insulin resistance and relative subunits with type 2 diabetes and related phenotypes. Of Tinsulin insufficiency. The final expression of the the seven genes that encode AMPK isoforms, we initially diabetic phenotype is strongly influenced by inheritance; chose PRKAA2, PRKAB1, and PRKAB2 because of their however, with the exception of rare monogenic forms of higher prior probability of association with type 2 diabetes, diabetes, common type 2 diabetes is thought to have a based on previous reports of genetic linkage, functional polygenic architecture (1). -

Identifying Novel Actionable Targets in Colon Cancer
biomedicines Review Identifying Novel Actionable Targets in Colon Cancer Maria Grazia Cerrito and Emanuela Grassilli * Department of Medicine and Surgery, University of Milano-Bicocca, Via Cadore 48, 20900 Monza, Italy; [email protected] * Correspondence: [email protected] Abstract: Colorectal cancer is the fourth cause of death from cancer worldwide, mainly due to the high incidence of drug-resistance toward classic chemotherapeutic and newly targeted drugs. In the last decade or so, the development of novel high-throughput approaches, both genome-wide and chemical, allowed the identification of novel actionable targets and the development of the relative specific inhibitors to be used either to re-sensitize drug-resistant tumors (in combination with chemotherapy) or to be synthetic lethal for tumors with specific oncogenic mutations. Finally, high- throughput screening using FDA-approved libraries of “known” drugs uncovered new therapeutic applications of drugs (used alone or in combination) that have been in the clinic for decades for treating non-cancerous diseases (re-positioning or re-purposing approach). Thus, several novel actionable targets have been identified and some of them are already being tested in clinical trials, indicating that high-throughput approaches, especially those involving drug re-positioning, may lead in a near future to significant improvement of the therapy for colon cancer patients, especially in the context of a personalized approach, i.e., in defined subgroups of patients whose tumors carry certain mutations. Keywords: colon cancer; drug resistance; target therapy; high-throughput screen; si/sh-RNA screen; CRISPR/Cas9 knockout screen; drug re-purposing; drug re-positioning Citation: Cerrito, M.G.; Grassilli, E. -

Dclk1+ Small Intestinal Epithelial Tuft Cells Display the Hallmarks of Quiescence and Self-Renewal
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 31 Dclk1+ small intestinal epithelial tuft cells display the hallmarks of quiescence and self-renewal Parthasarathy Chandrakesan1,2, Randal May1,3, Dongfeng Qu1,3, Nathaniel Weygant1, Vivian E. Taylor1, James D. Li1, Naushad Ali1,2, Sripathi M. Sureban1, Michael Qante4, Timothy C. Wang5, Michael S. Bronze1, Courtney W. Houchen1,2,3,6 1Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA 2Stephenson Oklahoma Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA 3Department of Veterans Affairs Medical Center, Oklahoma City, OK, USA 4Klinikum rechts der Isar, II. Medizinische Klinik, Technische Universität München, Munich, Germany 5Department of Digestive and Liver Diseases, Columbia University Medical Center, New York, NY, USA 6COARE Biotechnology, Oklahoma City, OK, USA Correspondence to: Courtney W. Houchen, e-mail: [email protected] Parthasarathy Chandrakesan, e-mail: [email protected] Keywords: Dclk1, self-renewal, pluripotency, quiescence Received: July 15, 2015 Accepted: August 19, 2015 Published: September 02, 2015 ABSTRACT To date, no discrete genetic signature has been defined for isolated Dclk1+ tuft cells within the small intestine. Furthermore, recent reports on the functional significance of Dclk1+ cells in the small intestine have been inconsistent. These cells have been proposed to be fully differentiated cells, reserve stem cells, and tumor stem cells. In order to elucidate the potential function of Dclk1+ cells, we FACS- sorted Dclk1+ cells from mouse small intestinal epithelium using transgenic mice expressing YFP under the control of the Dclk1 promoter (Dclk1-CreER;Rosa26-YFP). Analysis of sorted YFP+ cells demonstrated marked enrichment (~6000 fold) for Dclk1 mRNA compared with YFP− cells. -

A New Computational Approach to Evaluating Systemic Gene–Gene Interactions in a Pathway Affected by Drug LY294002
processes Article A New Computational Approach to Evaluating Systemic Gene–Gene Interactions in a Pathway Affected by Drug LY294002 Shinuk Kim College of Kyedang General Education, Sangmyung University, Cheonan 31066, Korea; [email protected]; Tel.: +82-(41)-550-5452; Fax: +82-(41)-550-5439 Received: 14 August 2020; Accepted: 23 September 2020; Published: 1 October 2020 Abstract: In this study, we investigate how drugs systemically affect genes via pathways by integrating information from interactions between chemical compounds and molecular expression datasets, and from pathway information such as gene sets using mathematical models. First, we adopt drug-induced gene expression datasets; then, employ gene set enrichment analysis tools for selecting candidate enrichment pathways; and lastly, implement the inverse algorithm package for identifying gene–gene regulatory networks in a pathway. We tested LY294002-induced datasets of the MCF7 breast cancer cell lines, and found a CELL CYCLE pathway with 101 genes, ERBB signaling pathway consisting of 82 genes, and MTOR pathway consisting of 45 genes. We consider two interactions: quantity strength depending on number of interactions, and quality strength depending on weight of interaction as positive (+) and negative ( ) interactions. Our methods revealed ANAPC1-CDK6 ( 0.412) and − − ORC2L- CHEK1(0.951) for the CELL CYCLE pathway; INS-RPS6 ( 3.125) and PRKAA2-PRKAA2 − (+1.319) for the MTOR pathway; and CBLB-RPS6KB1 ( 0.141), RPS6KB1-CBLC (+0.238) for the ERBB − signaling pathway to be top quality interactions. Top quantity interactions discovered include 12; the CDC ( ,+) gene family for the CELL CYCLE pathway, 20; PIK3 ( ), 23; PIK3CG (+) for the MTOR − − pathway, 11; PAK ( ), 10; PIK3 (+) for the ERBB signaling pathway. -

Association Study of AMP-Activated Protein Kinase Subunit Genes In
European Journal of Endocrinology (2009) 161 405–409 ISSN 0804-4643 CLINICAL STUDY Association study of AMP-activated protein kinase subunit genes in polycystic ovary syndrome Kari Sproul1,2, Michelle R Jones3, Ricardo Azziz1,2,4 and Mark O Goodarzi1,3,4,5 1Department of Obstetrics and Gynecology, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA, 2Department of Obstetrics and Gynecology, the David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA, 3Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, Room B-131, Los Angeles, California 90048, USA, 4Department of Medicine, the David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA and 5Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA (Correspondence should be addressed to M O Goodarzi at Division of Endocrinology, Diabetes and Metabolism, Department of Medicine, Cedars-Sinai Medical Center; Email: [email protected]) Abstract Objective: To examine the genes for AMP-activated protein kinase (AMPK) subunits a2(PRKAA2) and g3(PRKAG3) as candidates for polycystic ovary syndrome (PCOS) and its component traits. Design and methods: A total of 287 white PCOS women were recruited from the reproductive endocrinology clinic at the University of Alabama at Birmingham and 187 white control subjects were recruited from the surrounding community. Seven PRKAA2 single nucleotide polymorphisms (SNPs) and four PRKAG3 SNPs were genotyped in PCOS cases and controls. Genotyping and association analysis were performed at Cedars-Sinai Medical Center. Results: Nominal associations of PRKAA2 variants with insulin-related traits and the PRKAG3 Pro71Ala variant with PCOS were not statistically significant after multiple testing correction. -

Doublecotin-Like Kinase 1 Increases Chemoresistance of Colorectal Cancer Cells Through
bioRxiv preprint doi: https://doi.org/10.1101/517425; this version posted January 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Doublecotin-like kinase 1 increases chemoresistance of colorectal cancer cells through the anti-apoptosis pathway Lianna Li1*, Kierra Jones1, Hao Mei2# 1 Biology Department, Tougaloo College. 500 West County Line Road, Tougaloo MS 39174 2 Department of Data Science, University of Mississippi Medical Center. 2500 North State Street, Jackson, MS 39216 *Corresponding author: Lianna Li, email: [email protected] #Co-Corresponding author: Hao Mei, email: [email protected] Kierra Jones: [email protected] Running title: DCLK1 increases chemoresistance of CRC cells bioRxiv preprint doi: https://doi.org/10.1101/517425; this version posted January 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Colorectal cancer (CRC) is the third most common cancer diagnosed and the second leading cause of cancer-related deaths in the United States. About 50% of CRC patients relapsed after surgical resection and ultimately died of metastatic disease. Cancer stem cells (CSCs) are believed to be the primary reason for the recurrence of CRC. Specific stem cell marker, doublecortin-like kinase 1 (DCLK1) plays critical roles in initiating tumorigenesis, facilitating tumor progression, and promoting metastasis of CRC. -
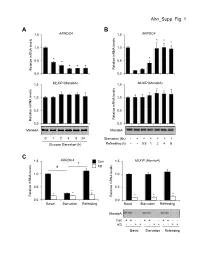
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

The Study of Copy Number Variations in the Regions of PRKAB2 and PPM1K Among Congenital Heart Defects Patients
The study of copy number variations in the regions of PRKAB2 and PPM1K among congenital heart defects patients Han-Quan Dong1 Yue-Xin Du2 1. Department of Pneumology, Tianjin Children’s Hospital, Tianjin, 300074–China 2. Department of Child Healthcare, Tianjin Municipal Women and Children health care center, Tianjin, 300070, China http://dx.doi.org/10.1590/1806-9282.65.6.786 SUMMARY OBJECTIVE: This study was to assess the genetic association of copy number variations in two genes (PRKAB2 and PPM1K) located in two regions (tetralogy of Fallot and ventricular septal defect) in a Chinese Han population. METHODS: A total of 200 congenital heart disease patients (100 tetralogy of Fallot patients and 100 ventricular septal defect patients) and 100 congenital heart defect-free controls were recruited, and quantitative real-time PCR analysis was used to replicate the asso- ciation of two copy number variations with congenital heart defects in a Chinese Han population. RESULTS: One deletion at PRKAB2 and one duplication at PPM1K were found in two of the tetralogy of Fallot patients, respectively; while all these regions were duplicated in both ventricular septal defect patients and in the 100 congenital heart defects-free controls. CONCLUSIONS: We replicated the copy number variations at the disease-candidate genes of PRKAB2 and PPM1K with tetralogy of Fallot in a Chinese Han population, and in patients with ventricular septal defect mutations in these two genes were not found. These results indicate the same molecular population genetics exist in these two genes with different ethnicity. This shows that these two genes are possibly specific pf tetralogy of Fallot candidates. -
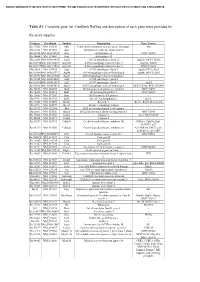
Table S1. Complete Gene List. Genbank Refseq and Description of Each Gene Were Provided By
Document downloaded from http://www.elsevier.es, day 24/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Table S1. Complete gene list. GenBank RefSeq and description of each gene were provided by the array supplier. Unigene GeneBank Symbol Description Gene Name/s Rn.11422 NM_033230 Akt1 V-akt murine thymoma viral oncogene homolog 1 Akt Rn.2104 NM_019288 App Amyloid beta (A4) precursor protein - Rn.23323 NM_001034933 Arsa Arylsulfatase A MGC125207 Rn.94004 NM_033443 Arsb Arylsulfatase B - Rn.6224 NM_001038495 Atg12 ATG12 autophagy related 12 Apg12l, MGC125080 Rn.101734NM_001108809 Atg16l1 ATG16 autophagy related 16-like 1 Apg16l, Wdr30 Rn.104199NM_001191560 Atg16l2 ATG16 autophagy related 16-like 2 RGD1311400 Rn.3084 NM_134394 Atg3 ATG3 autophagy related 3 Apg3l, PIG-1, Pig1 Rn.163086NM_001025711 Atg4b ATG4 autophagy related 4 homolog B Apg4b, MGC112887 Rn.23378 NM_001107948 Atg4c ATG4 autophagy related 4 homolog C - Rn.98385 NM_001014250 Atg5 ATG5 autophagy related 5 - Rn.162765NM_001012097 Atg7 ATG7 autophagy related 7 Apg7l Rn.35248 NM_001014218 Atg9a ATG9 autophagy related 9 homolog A MGC105908, RGD1310450 Rn.36696 NM_022698 Bad BCL2-associated agonist of cell death MGC72439 Rn.14598 NM_053812 Bak1 BCL2-antagonist/killer 1 MGC108627 Rn.10668 NM_017059 Bax Bcl2-associated X protein - Rn.9996 NM_016993 Bcl2 B-cell CLL/lymphoma 2 Bcl-2 Rn.10323 NM_031535 Bcl2l1 Bcl2-like 1 Bcl-xl, Bcl2l, Bclx, bcl-X Rn.2776 NM_053739 Becn1 Beclin 1, autophagy related - Rn.31142 NM_022684 -

(AMP)-Activated Protein Kinase: a New Target for Nutraceutical Compounds
International Journal of Molecular Sciences Review Adenosine Monophosphate (AMP)-Activated Protein Kinase: A New Target for Nutraceutical Compounds Fabiola Marín-Aguilar 1, Luis E. Pavillard 1, Francesca Giampieri 2, Pedro Bullón 1 and Mario D. Cordero 1,* 1 Research Laboratory, Oral Medicine Department, University of Sevilla, Sevilla 41009, Spain; [email protected] (F.M.-A.); [email protected] (L.E.P.); [email protected] (P.B.) 2 Dipartimento di Scienze Cliniche Specialistiche ed Odontostomatologiche—Sez. Biochimica, Università Politecnica delle Marche, Ancona 60100, Italy; [email protected] * Correspondence: [email protected]; Tel.: +34-954-481-120; Fax: +34-954-486-784 Academic Editor: Rosa M. Lamuela-Raventós Received: 17 November 2016; Accepted: 23 January 2017; Published: 29 January 2017 Abstract: Adenosine monophosphate-activated protein kinase (AMPK) is an important energy sensor which is activated by increases in adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio and/or adenosine diphosphate (ADP)/ATP ratio, and increases different metabolic pathways such as fatty acid oxidation, glucose transport and mitochondrial biogenesis. In this sense, AMPK maintains cellular energy homeostasis by induction of catabolism and inhibition of ATP-consuming biosynthetic pathways to preserve ATP levels. Several studies indicate a reduction of AMPK sensitivity to cellular stress during aging and this could impair the downstream signaling and the maintenance of the cellular energy balance and the stress resistance. However, several diseases have been related with an AMPK dysfunction. Alterations in AMPK signaling decrease mitochondrial biogenesis, increase cellular stress and induce inflammation, which are typical events of the aging process and have been associated to several pathological processes.