Santosh D. Bhosale D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fromcelebration Tocelebration
FromCelebration toCelebration Dress Code for Academic Events This guide introduces the dress code for academic events and festivities at Lappeenranta University of Technology. These festivities include the public defence of a dissertation, the post-doctoral karonkka banquet, and the conferment. Lappeenranta University of Technology was established in 1969 and, compared to many other universities, does not have long-standing traditions in academic festivities and especially doctoral conferment ceremonies. The following dress code should be observed at academic events at Lappeenranta University of Technology to establish in-house traditions. The instructions may, in some respects, differ from those of other universities. For instance with regard to colours worn by women, these instructions do not follow strict academic etiquette. 2 Contents Public Defence of a Dissertation 4 Conferment Ceremony 5 Dark Suit 6 Dark Suit, Accessories 8 Womens Semi-Formal Daytime Attire 10 White Tie 12 White Tie, Accessories 14 Womens Dark Suit, Doctoral Candidate 16 Womens Formal Daytime Attire, Doctor 18 Formal Evening Gown 20 Men's Informal Suit 22 Womens Informal Suit 24 Decorations 26 Doctoral Hat 28 Marshals 30 3 Public Defence of a Dissertation, Karonkka Banquet PUBLIC DEFENCE KARONKKA DOCTORAL CANDIDATE, White tie and tails, black vest White tie and tails, white vest if MALE (dark suit). ladies present (dark suit). Doctors: OPPONENT, doctoral hat. MALE CUSTOS, MALE p.12 p.12 DOCTORAL CANDIDATE, Womens dark suit, Formal evening gown, black. FEMALE high neckline, long sleeves, Doctors: doctoral hat. OPPONENT, suit with short skirt or trousers. FEMALE Opponent and custos: with CUSTOS, decorations. FEMALE p.16 p.20 CANDIDATES COMPANION, Semi-formal daytime attire, Formal evening gown, black. -
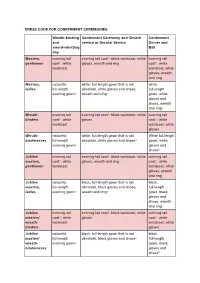
Dress Code Table
DRESS CODE FOR CONFERMENT CEREMONIES Wreathbinding Conferment Ceremony and Church Conferment and service or Secular Service Dinner and swordwhetting Ball day Masters, evening tail evening tail coat 1), black waistcoat, white evening tail gentlemen coat 1), white gloves, wreath and ring coat 1), white waistcoat waistcoat, white gloves, wreath and ring Masters, colourful, white, fulllength gown that is not white, ladies fulllength décolleté, white gloves and shoes, fulllength evening gown 2) wreath and ring 3) gown, white gloves and shoes, wreath and ring 5) Wreath evening tail evening tail coat 1), black waistcoat, white evening tail binders coat 1), white gloves coat 1), white waistcoat waistcoat, white gloves Wreath colourful, white, fulllength gown that is not White fulllength binderesses fulllength décolleté, white gloves and shoes 3) gown, white evening gown 2) gloves and shoes 5) Jubilee evening tail evening tail coat 1), black waistcoat, white evening tail masters, coat 1), white gloves, wreath and ring coat 1), white gentlemen waistcoat waistcoat, white gloves, wreath and ring Jubilee colourful, black, fulllength gown that is not black, masters, fulllength décolleté, black gloves and shoes, fulllength ladies evening gown 2) wreath and ring 4) gown, black gloves and shoes, wreath and ring 5) Jubilee evening tail evening tail coat 1), black waistcoat, white evening tail masters' coat 1), white gloves coat 1), white wreath waistcoat waistcoat, white binders gloves Jubilee colourful, black, fulllength gown that is not -

Promotio Ordinis Philosophorum Mmxvii
PROMOTIO ORDINIS PHILOSOPHORUM MMXVII Content 2 Dear Promovendus 4 Review of the matricula and delivering the diploma 5 Dance rehearsal 9 Program 15 Relatives participating in the Conferment Act 16 Dress codes 25 Academic Insignia 27 Conferment Artwork 27 Etiquette at the Conferment Ceremony 28 Photograhpy 33 Briefing for the promovendi 34 Contact information 1 Dear Promovendus, A warm welcome to the 97th Conferment Ceremony of the Faculty of Philosophy on 25th - 27th of May 2017! The registration is now closed, and the highlight of the spring is approaching day by day. You are holding the information package that has all the information necessary in prepa- ration for the Ceremony and also during the event itself. The purpose of these instructions is to assure that event runs smoothly and that each go smoothly and to make sure that. I would ask you to carefully read through the content of this booklet and follow the given instructions. If anything is still puzzling you, the Committee is happy to answer any questions. This information package contains detailed information on the schedule of the Conferment Ceremony and prac- tical tips for celebration. The booklet has been designed in such a form that it is also possible to carry it during the events. Some of the information will be specified further as the Conferment approaches and we hope that you follow the website and e-mail newsletters. In addition, the Conferment Committee organizes a Briefing session on 18th of April, 18.00 o’clock at the University Great Hall., You are warmly welcome there to hear about the Cere- mony and related issues and to ask your own questions. -

Academic Festivities
ACADEMIC FESTIVITIES A GUIDE FOR PARTICIPANTS TO THE DOCTORAL CONFERMENT CEREMONY AT THE UNIVERSITY OF EASTERN FINLAND, JOENSUU CAMPUS IN 2019 FOREWORD BY THE MASTER OF CEREMONIES ESTEEMED DOCTORS! It is my honour and pleasure to serve as the tial noise is becoming increasingly difficult. Master of Ceremonies for the third Doctor- Faced with complex questions, it is tempt- al Conferment Ceremony at the University ing to seek answers that are simple and easy of Eastern Finland. The Conferment Cere- – and often misleading. Now more than mony celebrates the value and significance ever, individuals must stand up to defend of academic education. The traditions of scientific knowledge. conferment and the festivities are rooted in the Middle Ages, and they have in many It is now time to celebrate the great aca- respects remained unchanged for centuries. demic journey that each of you new doctors have made over the course of your disser- The 2019 Conferment Ceremony will be- tations. The three-day festivities are held stow honorary doctorates to 12 scholars or in honour of the achievements of the new socially distinguished persons and confer honorary doctors and promovendi. doctoral degrees to 89 doctors from three faculties. This guide contains information and in- structions on the upcoming academic cele- Scientific knowledge, the most reliable form bration. A very warm welcome to the Doc- of information founded on evidence and toral Conferment Ceremony! research, finds itself hard-pressed in the rapidly changing modern world. Never be- fore -

Doctor of Holy Scripture’ Dr Stephen Pietsch
Ego etiam sum doctor scripturae: an historical and contemporary reflection on Luther’s life and work 145 Ego etiam sum doctor scripturae: an historical and contemporary reflection on Luther’s life and work as ‘doctor of holy scripture’ Dr Stephen Pietsch Stephen Pietsch is a pastoral theology lecturer at ALC, specialising in counselling and homilietics. Introduction ‘Doctor’ was arguably Luther’s most significant self-identification. Variants of the Latin phrase in the title of this essay, ‘I am also a doctor of scripture’, appear many times in Luther’s sermons and other writings, right through his career.1 It is the one designation which most closely captures Luther’s role in the three interconnected worlds he inhabited: university, local church and wider Reformation movement. He routinely signed his name ‘Doctor Martin Luther’ on letters and other documents, and in most of the documented references to Luther from his own time, even some of the unfavourable ones, he is named in the same way. Today’s egalitarian world refers to him simply by his name, but in the highly role-structured society of sixteenth century Germany, Luther was definitely ‘Herr Doktor’, even to most of his close associates and friends.2 It is a well-known part of the Luther story that he was awarded his doctoral degree at the University of Wittenberg in 1512. However, what is really known about this degree? How did Luther himself understand its value and importance? What did it mean in his world? How did Luther earn it? What role did it play in his reforming work? What insights from all this might be informative and even fruitful for us today, as Lutheran theological educators who stand on the threshold of the Reformation’s 500th anniversary and—at the same moment—look forward to a new millennium? Doctoral degrees in the 16th century In the early 16th century doctoral degrees were much rarer than they are today; they were a mark of high honour, and many important scholars were never awarded a doctoral degree. -

Skywatching in the Ancient World: New Perspectives in Cultural Astronomy—Studies in Honor of Anthony F
SKYWATCHING in the Ancient World MES OAM ERICAN W ORLDS From the Olmecs to the Danzantes GENERAL EDITORS: Davíd Carrasco and Eduardo Matos Moctezuma EDITORIAL BOARD: Michio Araki, Alfredo López Austin, Anthony Aveni, Elizabeth Boone, and Charles H. Long After Monte Albán: Transformation and Negotiation in Oaxaca, Mexico, JEFFREY P. BLOMSTER, EDITOR The Apotheosis of Janaab’ Pakal: Science, History, and Religion at Classic Maya Palenque, GERARDO ALDANA Commoner Ritual and Ideology in Ancient Mesoamerica, NANCY GONLIN AND JON C. LOHSE, EDITORS Eating Landscape: Aztec and European Occupation of Tlalocan, PHILIP P. ARNOLD Empires of Time: Calendars, Clocks, and Cultures, Revised Edition, ANTHONY AVENI Encounter with the Plumed Serpent: Drama and Power in the Heart of Mesoamerica, MAARTEN JANSEN AND GABINA AURORA PÉREZ JIMÉNEZ In the Realm of Nachan Kan: Postclassic Maya Archaeology at Laguna de On, Belize, MARILYN A. MASSON Invasion and Transformation: Interdisciplinary Perspectives on the Conquest of Mexico, REBECCA P. BRIENEN AND MARGARET A. JACKSON, EDITORS Life and Death in the Templo Mayor, EDUARDO MATOS MOCTEZUMA The Madrid Codex: New Approaches to Understanding an Ancient Maya Manuscript, GABRIELLE VAIL AND ANTHONY AVENI, EDITORS Mesoamerican Ritual Economy: Archaeological and Ethnological Perspectives, E. CHRISTIAN WELLS AND KARLA L. DAVIS-SALAZAR, EDITORS Mesoamerica’s Classic Heritage: Teotihuacan to the Aztecs, DAVÍD CARRASCO, LINDSAY JONES, AND SCOTT SESSIONS, EDITORS Mockeries and Metamorphoses of an Aztec God: Tezcatlipoca, “Lord of the Smoking Mirror,” GUILHEM OLIVIER, TRANSLATED BY MICHEL BESSON Rabinal Achi: A Fifteenth-Century Maya Dynastic Drama, ALAIN BRETON, EDITOR; TRANSLATED BY TERESA LAVENDER FAGAN AND ROBERT SCHNEIDER Representing Aztec Ritual: Performance, Text, and Image in the Work of Sahagún, ELOISE QUIÑONES KEBER, EDITOR Ruins of the Past: The Use and Perception of Abandoned Structures in the Maya Lowlands, TRAVIS W. -

What Is the Best Way to Begin Learning About Fashion, Trends, and Fashion Designers?
★ What is the best way to begin learning about fashion, trends, and fashion designers? Edit I know a bit, but not much. What are some ways to educate myself when it comes to fashion? Edit Comment • Share (1) • Options Follow Question Promote Question Related Questions • Fashion and Style : Apart from attending formal classes, what are some of the ways for someone interested in fashion designing to learn it as ... (continue) • Fashion and Style : How did the fashion trend of wearing white shoes/sneakers begin? • What's the best way of learning about the business behind the fashion industry? • Fashion and Style : What are the best ways for a new fashion designer to attract customers? • What are good ways to learn more about the fashion industry? More Related Questions Share Question Twitter Facebook LinkedIn Question Stats • Latest activity 11 Mar • This question has 1 monitor with 351833 topic followers. 4627 people have viewed this question. • 39 people are following this question. • 11 Answers Ask to Answer Yolanda Paez Charneco Add Bio • Make Anonymous Add your answer, or answer later. Kathryn Finney, "Oprah of the Internet" . One of the ... (more) 4 votes by Francisco Ceruti, Marie Stein, Unsah Malik, and Natasha Kazachenko Actually celebrities are usually the sign that a trend is nearing it's end and by the time most trends hit magazine like Vogue, they're on the way out. The best way to discover and follow fashion trends is to do one of three things: 1. Order a Subscription to Women's Wear Daily. This is the industry trade paper and has a lot of details on what's happen in fashion from both a trend and business level. -

Martin Kjellgren
MARTIN KJELLGREN sEKELBOKFÖRLAG his doctoral thesis. Taming the Prophets Astrology, Orthodoxy and the W ord of God in Early Modern Sweden Taming the Prophets Astrology, Orthodoxy and the W ord of God in Early Modern Sweden Martin Kjellgren SEKEL Denna bok publiceras med stöd av Kungl. Gustav Adolfs Akademien för svensk folkkultur Nationella forskarskolan i historia © Sekel Bokförlag och författaren, Lund 2orr Omslag: Johanna Åkerberg Omslagsillustration: Quae sup ra nos, nihil ad nos, emblem från Andrea Alciati, Emblemata, Lyon r 550 Engelsk språkgranskare: Deirdre Moore Korrekturläsare: Anneli Collins, Mikaellsacson Grafisk form: I&J Tryck: lnterPress, Budapest 2orr ISBN 978-91-85767-87-8 Contents Acknowledgements 7 PROLOGUE At the End ofTime II CHAPTER ONE Introduction: Between Nestor and Prometheus 19 CHAPTER TWO Astrology in the Northlands 53 CHAPTER THREE Piloting the Wreck of St. Peter CHAPTER FOUR Monopolizing Prophecy 129 CHAPTER FIVE The Reluctant Dissident CHAPTER SIX Ruling the Last Days 241 CHAPTER SEVEN Taming the Prophets Summary References Index nominum Index rerum Acknowledgements To every thing there is a season, and a time to every purpose under the heaven: A time to be born, and a time to die; a time to plant, and a time to pluck up that which is planted; A time to kil!, and a time to heal; a time to break down, and a time to build up ... A time to love, and a time to hate; a time of war, and a time of peace. Ecclesiastes 3:1-3, 8. uring my doctoral studies, thetimeshave been shifting. There have Dbeen times of extreme weariness. -

Community-University Research Partnerships: Reflections on the Canadian Social Economy Experience
Community-University Research Partnerships: Reflections on the Canadian Social Economy Experience Community-University Research Partnerships: Reflections on the Canadian Social Economy Experience Peter V. Hall and Ian MacPherson, Editors © 2011 University of Victoria © 2011 University of Victoria The moral rights of the authors are asserted. Published by University of Victoria, 3800 Finnerty Road, Victoria, BC V8P 5C2 ISBN 978-1-55058-449-3 (print) ISBN 978-1-55058-450-9 (PDF) ISBN 978-1-55058-451-6 (epub) Printed and bound in Canada by University of Victoria Book design by Kold Design Library and Archives Canada Cataloguing in Publication Community-university research partnerships : reflections on the Canadian social economy experience / Peter V. Hall and Ian MacPherson, editors. Includes bibliographical references. Issued also in electronic formats. ISBN 978-1-55058-449-3 1. Canadian Social Economy Research Partnerships. 2. Community and college--Canada. 3. Education, Higher-- Research--Canada. 4. Communities--Research--Canada. I. Hall, P. V. (Peter V.), 1968- II. MacPherson, Ian, 1939- LC238.4.C3C66 2011 378.1’03 C2011-907901-1 Research for this book was funded by the Social Sciences and Humanities Research Council of Canada and the University of Victoria. Canadian Social Economy Hub www.socialeconomyhub.ca This publication is licensed under a Creative Commons License, Attribution- Noncommercial-No Derivative 3.0 Unported Canada: see www.creativecommons. org. The text may be reproduced for non-commercial purposes, provided that credit -
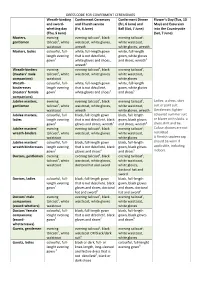
DRESS CODE for CONFERMENT CEREMONIES Wreath-Binding And
DRESS CODE FOR CONFERMENT CEREMONIES Wreath-binding Conferment Ceremony Conferment Dinner Flower’s Day (Tue, 13 and sword- and Church service (Fri, 6 June) and May) and Excursion whetting day (Fri, 6 June) Ball (Sat, 7 June) into the Countryside (Thu, 5 June) (Sat, 7 June) Masters, evening evening tailcoat1, black evening tailcoat1, gentlemen tailcoat1, white waistcoat, white gloves, white waistcoat, waistcoat wreath white gloves, wreath Masters, ladies colourful, full- white, full-length gown white, full-length length evening that is not décolleté, gown, white gloves gown2 white gloves and shoes, and shoes, wreath5 wreath3 Wreath-binders evening evening tailcoat1, black evening tailcoat1, (masters’ male tailcoat1, white waistcoat, white gloves white waistcoat, companions) waistcoat white gloves Wreath- colourful, full- white, full-length gown white, full-length binderesses length evening that is not décolleté, gown, white gloves (masters’ female gown2 white gloves and shoes3 and shoes5 companions) Jubilee masters, evening evening tailcoat1, black evening tailcoat1, Ladies: a dress, skirt gentlemen tailcoat1, white waistcoat, white gloves, white waistcoat, suit or pant suit. waistcoat wreath white gloves, wreath Gentlemen: lighter- Jubilee masters, colourful, full- black, full-length gown black, full-length coloured summer suit ladies length evening that is not décolleté, black gown, black gloves or blazer with slacks, a gown2 gloves and shoes, wreath4 and shoes, wreath5 dress shirt and tie. Jubilee masters’ evening evening tailcoat1, black evening tailcoat1, Colour choices are not wreath-binders tailcoat1, white waistcoat, white gloves white waistcoat, restricted. waistcoat white gloves A Finnish student cap Jubilee masters’ colourful, full- black, full-length gown black, full-length should be worn if wreath binderesses length evening that is not décolleté, black gown, black gloves applicable, including gown2 gloves and shoes4 and shoes5 indoors. -

Academical Dress in Sweden
Transactions of the Burgon Society Volume 13 Article 3 1-1-2013 Academical Dress in Sweden Klaus Solberg Søilen Halmstad University Follow this and additional works at: https://newprairiepress.org/burgonsociety Recommended Citation Søilen, Klaus Solberg (2014) "Academical Dress in Sweden," Transactions of the Burgon Society: Vol. 13. https://doi.org/10.4148/2475-7799.1107 This Article is brought to you for free and open access by New Prairie Press. It has been accepted for inclusion in Transactions of the Burgon Society by an authorized administrator of New Prairie Press. For more information, please contact [email protected]. Transactions of the Burgon Society, 13 (2013), pages 28–38 Academical Dress in Sweden By Klaus Solberg Søilen f there is little written on the subject of academical dress in Sweden it is because it has never played a major role in academic ceremonies in this country, or for that matter Iin any of the Nordic countries.1 We are reminded of this for example when comparing Swedish academical dress to the colourful displays of the robes and gowns in Anglo-Saxon countries. Instead the history of Swedish academical dress is primarily a story about in- signia, beginning with what to wear on one’s head. The insignia used in Swedish academic ceremonies are, besides the hat, the laurel wreath, the ring and the diploma. Of these it is the laurel wreath which is particular to the Swedish system, recalling the many portraits depicting Dante Alighieri.2 Of course the tradition of the laurel wreath is much older than Renaissance Italy, going back to Greek mythology. -

Information About the Conferment Ceremony for New Doctors During 2010/2011 at Stockholm University
Information about the Conferment Ceremony for New Doctors during 2010/2011 at Stockholm University. Updated: 2010-09-27 Stockholm University will arrange a conferment ceremony for new doctors and inauguration of new professors on Friday 30 September 2011. The ceremony will take place at 5 p.m. in the Blue Hall of the Stockholm City Hall and will be followed by a banquet in the Gold Hall, with dancing in the Blue Hall. Invitation Those doctorates who have defended their doctoral thesis during 2010/2011 and who have received their doctoral degree, will receive an invitation to the ceremony in the middle of June 2011. The International Student Office is responsible for issuing the degree. If you have questions about your degree please contact their office. If you applied for your degree after the end of the term it is necessary to contact us if you wish to participate in the conferment ceremony. Registration will be made on www.su.se/doktorspromotion according to the information given on the invitation card. Persons who are not able to participate in the 2011 ceremony may be conferred a doctoral degree next year, in 2012. Please notify this in writing to the Event Manager, no later than 30 April 2012. This may be done by sending an e-mail to: [email protected] or by posting a letter to Stockholms universitet, Konferensservice, SE-106 91 Stockholm, Sweden. Insignia presented during the ceremony During the ceremony proceedings you will be presented with a doctoral hat if you are a lawyer or a laurel if you are from one of the other faculties.