How Being Synanthropic Affects the Gut Bacteriome and Mycobiome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
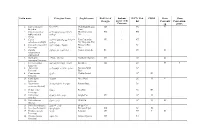
Red List of Endemic IUCN Red CITES Bern Bonn Georgia Species of the List Conventi Convention Caucasus on (CMS) 1
Latin name Georgian Name English name Red List of Endemic IUCN Red CITES Bern Bonn Georgia species of the list Conventi Convention Caucasus on (CMS) 1. Capra aegagrus niamori Wild Goat, Bezoar CR VU II Erxleben. Goat 2. Capra caucasica dasavleTkavkasiuri West Caucasian EN + EN Güldenstädt & jixvi Tur Pallas. 3. Capra aRmosavleTkavkasiuri East Caucasian VU + NT cylindricornis Blyth. jixvi Tur, Dagestan Tur 4. Capreolus capreolus evropuli Sveli European Roe LC Linnaeus. Deer 5. Gazella qurciki, jeirani Goitered Gazelle RE VU II subgutturosa Güldenstädt. 6. Rupicapra arCvi, fsiti Northern Chamois EN LC II rupicapra Linnaeus. 7. Cervus elaphus keTilSobili iremi Red Deer CR LC II I Linnaeus. 8. Sus scrofa gareuli Rori, taxi Eurasian Wild LC Linnaeus. Boar 9. Canis aureus tura Golden Jackal LC III Linnaeus. 10. Canis lupus mgeli Grey Wolf LC II II Linnaeus. 11. Nyctereutes enotisebri ZaRli Racoon Dog LC procyonoides Gray. 12. Vulpes vulpes mela Red Fox LC III Linnaeus. 13. Felis chaus lelianis kata Jungle Cat VU LC II Schreber. 14. Felis silvestris tyis kata Wild Cat LC II II Shreber. 15. Felis libyca Forster. velis kata Steppe Cat 16. Lynx lynx Linnaeus. focxveri Eurasian Lynx CR LC II 17. Panthera pardus jiqi Leopard CR NT I II Linnaeus. 18. Hyaena hyaena afTari Striped Hyaena CR NT Linnaeus. 19. Lutra lutra wavi Eurasian Otter, VU NT I II Linnaeus. Common Otter 20. Martes foina kldis kverna Stone Marten, LC III Erxleben. Beech Marten 21. Martes martes tyis kverna European Pine LC Linnaeus. Marten 22. Meles meles maCvi Eurasian Badger LC Linnaeus. 23. Mustela lutreola waula European Mink EN II Linnaeus. -

Mammal Survey Pol'ana (Slovakia) 2005 5�
MAMMAL SURVEY POL 'ANA (S LOVAKIA ) 2005 RESULTS OF TEN DAYS ’ FIELD WORK RAPPORT 2009.23 Juli 2009 Uitgave van de Veldwerkgroep van de Zoogdiervereniging MAMMAL SURVEY POL 'ANA (S LOVAKIA ) 2005 RESULTS OF TEN DAYS ’ FIELD WORK Editors Jan Buys Jeroen Willemsen Translations Karin de Bie Authors Jan Boshamer Jan Buys Annemarie van Diepenbeek Rob Koelman Jeroen van der Kooij Rudy van der Kuil Kees Mostert Janny Resoort Froukje Rienks Kamiel Spoelstra Anke van der Wal Jeroen Willemsen Illustration on front cover Froukje Rienks Photographs Jan Boshamer Jan Buys Jeroen van der Kooij Rudy van der Kuil Janny Resoort Kamiel Spoelstra Joost Verbeek Anke van der Wal Uitgave van de Veldwerkgroep van de Zoogdiervereniging Rapport 2009.23 Arnhem, juli 2009 ISBN: 978-90-79924-12-7 All publications of the Veldwerkgroep can be downloaded free of charge from our website: http://www.zoogdiervereniging.nl/node/137 The English page of the website contains all publications in English: http://www.zoogdiervereniging.nl/node/411 SUMMARY by: Jan Buys and Jeroen Willemsen From July 27th until August 4th 2005, the Veldwerkgroep (VWG) of the Dutch Mammal Society (Zoogdiervereniging) paid a visit to the Pol’ana Biosphere reserve in Slovakia. A workshop in the summer is a traditional part of the VWG’s yearly program, aimed at surveying mammal species which are rare or absent in the Netherlands, and to extend and exchange knowledge of survey methods and species. Secondly, these workshops abroad aim to enlarge the knowledge of presence and abundance of mammals, and to a lesser extent of other fauna groups, in the area visited. -

Revised and Commented Checklist of Mammal Species of the Romanian Fauna
REVISED AND COMMENTED CHECKLIST OF MAMMAL SPECIES OF THE ROMANIAN FAUNA DUMITRU MURARIU Abstract. Due to the permanent influences of different factors (habitat degradation andfragmentation, deforestation, infrastructure and urbanization, natural extension or decreasing of some species’ distribution, increasing number of alien species etc.), from time to time the faunistic structure of a certain area is changing. As a result of the permanent and increasing anthropic and invasive species’ pressure, our previous checklist of recent mammals from Romania (since 1984) became out of date. A number of 108 taxa are mentioned in this checklist, representing 7 orders of mammals: Insectivora (10 species), Chiroptera (30 sp.), Lagomorpha (2 sp.), Rodentia (35 sp.), Cetacea (3 sp.), Carnivora (19 sp.), Artiodactyla (8 sp.). In this list are mentioned the scientific and vernacular names (in Romanian and English languages), species distribution and conservation status, according to the Romanian regulations. Thus, only 21 species have stable populations while 76 have populations in decline or in drastic decline. Other categories are not evaluated or even present an increase in their population. Key words: species and subspecies, recent mammals, distribution, conservation. 1. INTRODUCTION MURARIU published ‘La Liste de Mammifère actuels de Roumanie; noms scientifiques et Roumains’ in 1984. That moment represented an astonishing progress in the knowledge of mammals, only in two decades. A number of 3,500 species (belonging to 1,000 genera) were reported by DAVID & GOLLEY (1965) and 4,170 species were reported by HONACKI et al. (1982). Description of new mammal species is continuing, and WILSON & REEDER DEEANN (2005) presented “... the taxonomic classification and distribution of the more than 5,400 species of mammals that exist today”. -

Seasonal Changes in Tawny Owl (Strix Aluco) Diet in an Oak Forest in Eastern Ukraine
Turkish Journal of Zoology Turk J Zool (2017) 41: 130-137 http://journals.tubitak.gov.tr/zoology/ © TÜBİTAK Research Article doi:10.3906/zoo-1509-43 Seasonal changes in Tawny Owl (Strix aluco) diet in an oak forest in Eastern Ukraine 1, 2 Yehor YATSIUK *, Yuliya FILATOVA 1 National Park “Gomilshanski Lisy”, Kharkiv region, Ukraine 2 Department of Zoology and Animal Ecology, Faculty of Biology, V.N. Karazin Kharkiv National University, Kharkiv, Ukraine Received: 22.09.2015 Accepted/Published Online: 25.04.2016 Final Version: 25.01.2017 Abstract: We analyzed seasonal changes in Tawny Owl (Strix aluco) diet in a broadleaved forest in Eastern Ukraine over 6 years (2007– 2012). Annual seasons were divided as follows: December–mid-April, April–June, July–early October, and late October–November. In total, 1648 pellets were analyzed. The most important prey was the bank vole (Myodes glareolus) (41.9%), but the yellow-necked mouse (Apodemus flavicollis) (17.8%) dominated in some seasons. According to trapping results, the bank vole was the most abundant rodent species in the study region. The most diverse diet was in late spring and early summer. Small forest mammals constituted the dominant group in all seasons, but in spring and summer their share fell due to the inclusion of birds and the common spadefoot (Pelobates fuscus). Diet was similar in late autumn, before the establishment of snow cover, and in winter. The relative representation of species associated with open spaces increased in winter, especially in years with deep snow cover, which may indicate seasonal changes in the hunting habitats of the Tawny Owl. -

A Review of the Results Obtained During the Field Study Group Summer Camps of the Dutch Mammal Society, 1986-2014
A review of the results obtained during the Field Study Group summer camps of the Dutch Mammal Society, 1986-2014 Jan Piet Bekker1, Kees Mostert2, Jan P.C. Boshamer3 & Eric Thomassen4 1Zwanenlaan 10, NL-4351 RX Veere, the Netherlands, e-mail: [email protected] 2Palamedesstraat 74, NL-2612 XS Delft, the Netherlands 3Vogelzand 4250, NL-1788 MP Den Helder, the Netherlands 4Middelstegracht 28a, NL-2312 TX Leiden, the Netherlands Abstract: The 28 summer camps of the Field Study Group of the Dutch Mammal Society organised between 1986 and 2014 are reviewed here. Over time the Field Study Group gradually spread out its activities throughout Europe, including former Eastern Bloc countries. Camp locations were found through contacts in host countries, who also assist in the preparation of camp activities. Out of a total of 160 participants from the Netherlands and Belgium, 80 attended a summer camp once and 80 joined more than once; 116 participants from local origin were active dur- ing these camps. For the 128 mammal species found, the observation techniques used are described. Overall, 7,662 small mammals were caught with live-traps and 990 bats were caught in mist nets. Among the trapped mammals, 421 casualties were counted, predominantly common and pygmy shrews in northern European countries. In pellets, pre- dominantly from barn owls, 21,620 small mammals were found. With detectors, 3,908 bats could be identified. Caves and (old) buildings were explored for bats, and the results of these surveys made up a large part of the total number of bats found. Sightings (> 1,740) and tracks & signs (> 1,194) revealed most of all the presence of carnivora and even- toed ungulates (Artiodactyla). -

Food Supply (Orthoptera, Mantodea, Rodentia and Eulipotyphla)
Slovak Raptor Journal 2017, 11: 1–14. DOI: 10.1515/srj-2017-0005. © Raptor Protection ofSlovakia (RPS) Food supply (Orthoptera, Mantodea, Rodentia and Eulipotyphla) and food preferences of the red-footed falcon (Falco vespertinus) in Slovakia Potravná ponuka (Orthoptera, Mantodea, Rodentia a Eulipotyphla) a potravné preferencie sokola kobcovitého (Falco vespertinus) na Slovensku Anton KRIŠTÍN, Filip TULIS, Peter KLIMANT, Kristián BACSA & Michal AMBROS Abstract: Food supply in the nesting territories of species has a key role to the species diet composition and their breeding success. Red-footed falcon (Falco vespertinus) preys predominantly on larger insect species with a supplementary portion of smaller vertebrates. In the breeding periods 2014 and 2016 their food supply, focusing on Orthoptera, Mantodea, Rodentia and Eulipotyphla, was analysed at five historical nesting sites of the species in Slovakia. Preference for these prey groups in the diet was also studied at the last active nesting site in this country. Overall we recorded 45 Orthoptera species (of which 23 species are known as the food of the red-footed falcon), one species of Mantodea, 10 species of Rodentia (of which 2 species are known as the food of the red-footed falcon) and 5 species of the Eulipotyphla order in the food supply. With regard to the availability of the falcons' preferred food, in both years the most suitable was the Tvrdošovce site, which continuously showed the greatest range and abundance of particular species. In the interannual comparison the insects showed lower variability in abundance than the small mammals. In 2014 the growth of the common vole (Microtus arvalis) population culminated and with the exception of a single site (Bodza) a slump in abundance was recorded in 2016. -

Martin Pfeiffer-Habilitation
Structuring of animal communities: Interspecific interactions and habitat selection among ants and small mammals Habilitationsschrift zur Erlangung der Venia Legendi an der Universität Ulm Fakultät für Naturwissenschaften vorgelegt von Dr. Martin Pfeiffer Ulm, Oktober 2007 Year’s end — Still in straw hat And sandals. Basho (1644 -1694) In dieser Arbeit werden die Untersuchungen zur Gemeinschaftsökologie von Ameisen und Kleinsäugern vorgestellt, die ich zwischen 1997 und 2007 durchgeführt oder betreut habe. Ich versichere, dass ich die vorliegende Arbeit ohne fremde Hilfe angefertigt und mich keiner anderen als der ausdrücklich angegebenen Hilfsmittel bedient habe. Martin Pfeiffer Ulm, 30. Oktober 2007 CONTENTS Acknowledgments 1 1. Disentangling life histories, organization, and functions in animal communities of tropical rainforests and arid areas – an overview 3 2. Internet-based ant taxonomy and biodiversity informatics 8 3. Ant diversity gradients and faunistic inventory 14 4. Null model studies of interspecific interactions: community structure of Malaysian ants 18 5. The Sarawak soil ant project: Niches, trophic levels, and community patterns in rainforest ants 21 6. Ant- plant mutualism: Myrmecochory - seed dispersal by ants 24 7. Spatial organization in Bornean small mammal assemblages 27 8. Rainforest logging in Borneo: impacts on non-volant small mammal assemblages 30 References 34 Research articles ordered Research articles belonging to Chapter 3 43 Pfeiffer M, Chimedregzen L, Ulykpan K (2003) Community organization and species richness of ants (Hymenoptera/Formicidae) in Mongolia along an ecological gradient from steppe to Gobi desert. Journal of Biogeography 30:1921-1935 Pfeiffer M, Schultz R, Radchenko A, Yamane S, Woyciechowski M, Ulykpan A, Seifert B (2006) A critical checklist of the ants of Mongolia (Hymenoptera : Formicidae). -

Isolation, Characterization, and Evolutionary Divergence of Mouse Rnase 6: Evidence for Unusual Evolution in Rodents
J Mol Evol (2004) 59:657–665 DOI: 10.1007/s00239-004-2657-0 Isolation, Characterization, and Evolutionary Divergence of Mouse RNase 6: Evidence for Unusual Evolution in Rodents Kimberly D. Dyer,1 Helene F. Rosenberg,1 Jianzhi Zhang2 1 Eosinophil Biology Section, Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA 2 Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor MI 48109, USA Received: 26 February 2004 / Accepted: 7 June 2004 [Reviewing Editor: Dr. Lauren Ancel Meyers] Abstract. The evolution of the ribonuclease A Key words: Ribonuclease — RNase A superfamily (RNase A) vertebrate-specific enzyme family is inter- — RNase k6 — RNase 6 — Host defense esting in that specific gene lineages appear to be responding to unique selective pressures in wildly diverse manners to generate proteins that are capable Introduction of reducing the infectivity of viruses, killing systemic pathogens, and inducing the growth of blood vessels all Ribonuclease 6 is a member of the ribonuclease A while maintaining the signature motifs of a ribonu- (RNase A) superfamily, which is believed to be the clease. In this paper, we present the DNA sequence and only enzyme family that is limited to vertebrates gene structure of Mus musculus RNase 6 and examine (Lander et al. 2001). This family has been well char- the expression pattern and enzymatic activity of the acterized structurally, with most members having recombinant protein. M. musculus RNase 6 has a eight cysteines as well as the lysine and the two his- limited expression pattern compared to human RNase tidines that form the catalytic triad (Beintema et al. -

List of Taxa for Which MIL Has Images
LIST OF 27 ORDERS, 163 FAMILIES, 887 GENERA, AND 2064 SPECIES IN MAMMAL IMAGES LIBRARY 31 JULY 2021 AFROSORICIDA (9 genera, 12 species) CHRYSOCHLORIDAE - golden moles 1. Amblysomus hottentotus - Hottentot Golden Mole 2. Chrysospalax villosus - Rough-haired Golden Mole 3. Eremitalpa granti - Grant’s Golden Mole TENRECIDAE - tenrecs 1. Echinops telfairi - Lesser Hedgehog Tenrec 2. Hemicentetes semispinosus - Lowland Streaked Tenrec 3. Microgale cf. longicaudata - Lesser Long-tailed Shrew Tenrec 4. Microgale cowani - Cowan’s Shrew Tenrec 5. Microgale mergulus - Web-footed Tenrec 6. Nesogale cf. talazaci - Talazac’s Shrew Tenrec 7. Nesogale dobsoni - Dobson’s Shrew Tenrec 8. Setifer setosus - Greater Hedgehog Tenrec 9. Tenrec ecaudatus - Tailless Tenrec ARTIODACTYLA (127 genera, 308 species) ANTILOCAPRIDAE - pronghorns Antilocapra americana - Pronghorn BALAENIDAE - bowheads and right whales 1. Balaena mysticetus – Bowhead Whale 2. Eubalaena australis - Southern Right Whale 3. Eubalaena glacialis – North Atlantic Right Whale 4. Eubalaena japonica - North Pacific Right Whale BALAENOPTERIDAE -rorqual whales 1. Balaenoptera acutorostrata – Common Minke Whale 2. Balaenoptera borealis - Sei Whale 3. Balaenoptera brydei – Bryde’s Whale 4. Balaenoptera musculus - Blue Whale 5. Balaenoptera physalus - Fin Whale 6. Balaenoptera ricei - Rice’s Whale 7. Eschrichtius robustus - Gray Whale 8. Megaptera novaeangliae - Humpback Whale BOVIDAE (54 genera) - cattle, sheep, goats, and antelopes 1. Addax nasomaculatus - Addax 2. Aepyceros melampus - Common Impala 3. Aepyceros petersi - Black-faced Impala 4. Alcelaphus caama - Red Hartebeest 5. Alcelaphus cokii - Kongoni (Coke’s Hartebeest) 6. Alcelaphus lelwel - Lelwel Hartebeest 7. Alcelaphus swaynei - Swayne’s Hartebeest 8. Ammelaphus australis - Southern Lesser Kudu 9. Ammelaphus imberbis - Northern Lesser Kudu 10. Ammodorcas clarkei - Dibatag 11. Ammotragus lervia - Aoudad (Barbary Sheep) 12. -

Identifing Priority Ecoregions for Rodent Conservation at the Genus Level
Oryx Vol 35 No 2 April 2001 Short Communication Identifing priority ecoregions for rodent conservation at the genus level Giovanni Amori and Spartaco Gippoliti Abstract Rodents account for 40 per cent of living high number of genera) 'threat-spots' for rodent conser- mammal species. Nevertheless, despite an increased vation. A few regions, mainly drylands, are singled out interest in biodiversity conservation and their high as important areas for rodent conservation but are not species richness, Rodentia are often neglected by con- generally recognized in global biodiversity assessments. servationists. We attempt for the first time a world-wide These are the remaining forests of Togo, extreme evaluation of rodent conservation priorities at the genus 'western Sahel', the Turanian and Mongolian-Manchu- level. Given the low popularity of the order, we rian steppes and the desert of the Horn of Africa. considered it desirable to discuss identified priorities Resources for conservation must be allocated first to within the framework of established biodiversity prior- recognized threat spots and to those restricted-range ity areas of the world. Two families and 62 genera are genera which may depend on species-specific strategies recognized as threatened. Our analyses highlight the for their survival. Philippines, New Guinea, Sulawesi, the Caribbean, China temperate forests and the Atlantic Forest of Keywords Biodiversity, conservation priorities, south-eastern Brazil as the most important (for their rodents, threatened genera, world ecoregions. Conservation efforts for rodents must be included in Introduction the general framework of mammalian diversity conser- With 26-32 recognized extant families and more than vation, focusing on a biodiversity/area approach. -

CICUAL; Approval Number C.FUA 14-023
Genomics of the capybara, two emblematic Colombian species María José Gómez-Hughes¹, Santiago Herrera-Álvarez1,2, Andrew J. Crawford¹ ¹Department of Biological Sciences, Universidad de los Andes, Bogotá, 111711, Colombia. ²Department of Ecology and Evolution, University of Chicago, Chicago, IL 60637, USA. Abstract Capybaras, which are native to South America, are not only the largest rodents in the world, but they also have a number of other characteristics that make them unique. They are semi-aquatic, grazing mammals and live in large groups where females engage in communal breeding. Males communally defend the territory through scent-marking with a specialized gland called the morillo and with two anal glands. Here we present the first genome assembly and annotation for the lesser capybara, Hydrochoerus isthmius, as well as the first transcriptome assembly for the capybara, H. hydrochaeris, both of which are comparable in completeness with previously published rodent genomes, and compared them with the previously published genome assembly for the capybara. We found evidence of reduction on the effective population size of both species, as well as big regions of genomic rearrangement with the guinea pig. Our phylogenetic analysis is consistent with previous phylogenies reported for the suborder Hystrichomorpha, but species related there is evidence for the capybara being a paraphyletic species. We hope that this study contributes for conservation efforts on these species, as well as a better understanding of all the characteristics that make them unique. Resumen Los chigüiros, nativos a América del Sur, no solamente son los roedores más grandes del mundo sino también tienen otras características que los hacen únicos. -

HANDBOOK of the MAMMALS of the WORLD Families of Volume 1: Carnivores
HANDBOOK OF THE MAMMALS OF THE WORLD Families of Volume 1: Carnivores Family Family English Subfamily Group name Species Genera Scientific name name number African Palm NANDINIIDAE 1 species Nandinia Civet Neofelis Pantherinae Big Cats 7 species Panthera Pardofelis Catopuma FELIDAE Cats Leptailurus Profelis Caracal Leopardus Felinae Small Cats 30 species Lynx Acinonyx Puma Otocolobus Prionailurus Felis PRIONODONTIDAE Linsangs 2 species Prionodon Viverricula Viverrinae Terrestrial Civets 6 species Civettictis Viverra Poiana Genettinae Genets and Oyans 17 species Genetta Civets, Genets VIVERRIDAE and Oyans Arctogalidia Macrogalidia Palm Civets and Paradoxurinae 7 species Arctictis Binturong Paguma Paradoxurus Cynogale Palm Civets and Chrotogale Hemigalinae 4 species Otter Civet Hemigalus Diplogale Family Family English Subfamily Group name Species Genera Scientific name name number Protelinae Aardwolf 1 species Proteles HYAENIDAE Hyenas Crocuta Bone-cracking Hyaeninae 3 species Hyaena Hyenas Parahyaena Atilax Xenogale Herpestes Cynictis Solitary Herpestinae 23 species Galerella Mongooses Ichneumia Paracynictis HERPESTIDAE Mongooses Bdeogale Rhynchogale Suricata Crossarchus Social Helogale Mungotinae 11 species Mongooses Dologale Liberiictis Mungos Civet-like Cryptoprocta Euplerinae Madagascar 3 species Eupleres Carnivores Fossa Madagascar EUPLERIDAE Carnivores Galidia Mongoose-like Galidictis Galidinae Madagascar 5 species Mungotictis Carnivores Salanoia Canis Cuon Lycaon Chrysocyon Speothos Cerdocyon CANIDAE Dogs 35 species Atelocynus Pseudalopex