Elementary Sequence Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
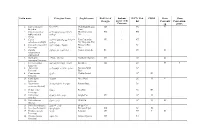
Red List of Endemic IUCN Red CITES Bern Bonn Georgia Species of the List Conventi Convention Caucasus on (CMS) 1
Latin name Georgian Name English name Red List of Endemic IUCN Red CITES Bern Bonn Georgia species of the list Conventi Convention Caucasus on (CMS) 1. Capra aegagrus niamori Wild Goat, Bezoar CR VU II Erxleben. Goat 2. Capra caucasica dasavleTkavkasiuri West Caucasian EN + EN Güldenstädt & jixvi Tur Pallas. 3. Capra aRmosavleTkavkasiuri East Caucasian VU + NT cylindricornis Blyth. jixvi Tur, Dagestan Tur 4. Capreolus capreolus evropuli Sveli European Roe LC Linnaeus. Deer 5. Gazella qurciki, jeirani Goitered Gazelle RE VU II subgutturosa Güldenstädt. 6. Rupicapra arCvi, fsiti Northern Chamois EN LC II rupicapra Linnaeus. 7. Cervus elaphus keTilSobili iremi Red Deer CR LC II I Linnaeus. 8. Sus scrofa gareuli Rori, taxi Eurasian Wild LC Linnaeus. Boar 9. Canis aureus tura Golden Jackal LC III Linnaeus. 10. Canis lupus mgeli Grey Wolf LC II II Linnaeus. 11. Nyctereutes enotisebri ZaRli Racoon Dog LC procyonoides Gray. 12. Vulpes vulpes mela Red Fox LC III Linnaeus. 13. Felis chaus lelianis kata Jungle Cat VU LC II Schreber. 14. Felis silvestris tyis kata Wild Cat LC II II Shreber. 15. Felis libyca Forster. velis kata Steppe Cat 16. Lynx lynx Linnaeus. focxveri Eurasian Lynx CR LC II 17. Panthera pardus jiqi Leopard CR NT I II Linnaeus. 18. Hyaena hyaena afTari Striped Hyaena CR NT Linnaeus. 19. Lutra lutra wavi Eurasian Otter, VU NT I II Linnaeus. Common Otter 20. Martes foina kldis kverna Stone Marten, LC III Erxleben. Beech Marten 21. Martes martes tyis kverna European Pine LC Linnaeus. Marten 22. Meles meles maCvi Eurasian Badger LC Linnaeus. 23. Mustela lutreola waula European Mink EN II Linnaeus. -

120421-24Recombschedule FINAL.Xlsx
Friday 20 April 18:00 20:00 REGISTRATION OPENS in Fira Palace 20:00 21:30 WELCOME RECEPTION in CaixaForum (access map) Saturday 21 April 8:00 8:50 REGISTRATION 8:50 9:00 Opening Remarks (Roderic GUIGÓ and Benny CHOR) Session 1. Chair: Roderic GUIGÓ (CRG, Barcelona ES) 9:00 10:00 Richard DURBIN The Wellcome Trust Sanger Institute, Hinxton UK "Computational analysis of population genome sequencing data" 10:00 10:20 44 Yaw-Ling Lin, Charles Ward and Steven Skiena Synthetic Sequence Design for Signal Location Search 10:20 10:40 62 Kai Song, Jie Ren, Zhiyuan Zhai, Xuemei Liu, Minghua Deng and Fengzhu Sun Alignment-Free Sequence Comparison Based on Next Generation Sequencing Reads 10:40 11:00 178 Yang Li, Hong-Mei Li, Paul Burns, Mark Borodovsky, Gene Robinson and Jian Ma TrueSight: Self-training Algorithm for Splice Junction Detection using RNA-seq 11:00 11:30 coffee break Session 2. Chair: Bonnie BERGER (MIT, Cambrige US) 11:30 11:50 139 Son Pham, Dmitry Antipov, Alexander Sirotkin, Glenn Tesler, Pavel Pevzner and Max Alekseyev PATH-SETS: A Novel Approach for Comprehensive Utilization of Mate-Pairs in Genome Assembly 11:50 12:10 171 Yan Huang, Yin Hu and Jinze Liu A Robust Method for Transcript Quantification with RNA-seq Data 12:10 12:30 120 Zhanyong Wang, Farhad Hormozdiari, Wen-Yun Yang, Eran Halperin and Eleazar Eskin CNVeM: Copy Number Variation detection Using Uncertainty of Read Mapping 12:30 12:50 205 Dmitri Pervouchine Evidence for widespread association of mammalian splicing and conserved long range RNA structures 12:50 13:10 169 Melissa Gymrek, David Golan, Saharon Rosset and Yaniv Erlich lobSTR: A Novel Pipeline for Short Tandem Repeats Profiling in Personal Genomes 13:10 13:30 217 Rory Stark Differential oestrogen receptor binding is associated with clinical outcome in breast cancer 13:30 15:00 lunch break Session 3. -

Python for Bioinformatics, Second Edition
PYTHON FOR BIOINFORMATICS SECOND EDITION CHAPMAN & HALL/CRC Mathematical and Computational Biology Series Aims and scope: This series aims to capture new developments and summarize what is known over the entire spectrum of mathematical and computational biology and medicine. It seeks to encourage the integration of mathematical, statistical, and computational methods into biology by publishing a broad range of textbooks, reference works, and handbooks. The titles included in the series are meant to appeal to students, researchers, and professionals in the mathematical, statistical and computational sciences, fundamental biology and bioengineering, as well as interdisciplinary researchers involved in the field. The inclusion of concrete examples and applications, and programming techniques and examples, is highly encouraged. Series Editors N. F. Britton Department of Mathematical Sciences University of Bath Xihong Lin Department of Biostatistics Harvard University Nicola Mulder University of Cape Town South Africa Maria Victoria Schneider European Bioinformatics Institute Mona Singh Department of Computer Science Princeton University Anna Tramontano Department of Physics University of Rome La Sapienza Proposals for the series should be submitted to one of the series editors above or directly to: CRC Press, Taylor & Francis Group 3 Park Square, Milton Park Abingdon, Oxfordshire OX14 4RN UK Published Titles An Introduction to Systems Biology: Statistical Methods for QTL Mapping Design Principles of Biological Circuits Zehua Chen Uri Alon -

Contributions to Biostatistics: Categorical Data Analysis, Data Modeling and Statistical Inference Mathieu Emily
Contributions to biostatistics: categorical data analysis, data modeling and statistical inference Mathieu Emily To cite this version: Mathieu Emily. Contributions to biostatistics: categorical data analysis, data modeling and statistical inference. Mathematics [math]. Université de Rennes 1, 2016. tel-01439264 HAL Id: tel-01439264 https://hal.archives-ouvertes.fr/tel-01439264 Submitted on 18 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. HABILITATION À DIRIGER DES RECHERCHES Université de Rennes 1 Contributions to biostatistics: categorical data analysis, data modeling and statistical inference Mathieu Emily November 15th, 2016 Jury: Christophe Ambroise Professeur, Université d’Evry Val d’Essonne, France Rapporteur Gérard Biau Professeur, Université Pierre et Marie Curie, France Président David Causeur Professeur, Agrocampus Ouest, France Examinateur Heather Cordell Professor, University of Newcastle upon Tyne, United Kingdom Rapporteur Jean-Michel Marin Professeur, Université de Montpellier, France Rapporteur Valérie Monbet Professeur, Université de Rennes 1, France Examinateur Korbinian Strimmer Professor, Imperial College London, United Kingdom Examinateur Jean-Philippe Vert Directeur de recherche, Mines ParisTech, France Examinateur Pour M.H.A.M. Remerciements En tout premier lieu je tiens à adresser mes remerciements à l’ensemble des membres du jury qui m’ont fait l’honneur d’évaluer mes travaux. -

Sequence Analysis Instructions
Sequence Analysis Instructions In order to predict your drug metabolizing phenotype from your CYP2D6 gene sequence, you must determine: 1) The assembled sequence from your two opposing sequencing reactions 2) If your PCR product even represents the human CYP2D6 gene, 3) The location of your sequence within the CYP2D6 gene, 4) Whether differences between your alleles and CYP2D6*1 sequence represents sequencing errors or polymorphisms in your sequence, and 5) The effect(s) of any polymorphisms on CYP2D6 protein sequence. As you analyze your gene sequence, copy and paste your analyses and results into a text file for your final lab report. If some factor, like the quality of your sequence, prevents you from carrying out the complete analysis, your grade will not be penalized—just complete as many of the steps below as possible, and include an explanation of why you could not complete the analysis in your final report. Font appearing in bold green italics describes questions to answer and items to include in your final report. Part 1: Assembling your CYP2D6 sequence from both directions The sequencing reaction only produces 700-800 bases of good sequence. To get most of the sequence of the 1.2 Kbp PCR product, sequencing reactions were performed from both directions on the PCR product. These need to be assembled into one sequence using the overlap of the two sequences. In order to assure that it is going in the forward direction, it is best to derive the reverse complement sequence from the reverse sequencing file. Take the text file and paste it into a program like http://www.bioinformatics.org/sms/rev_comp.html. -

Agenda: Gene Prediction by Cross-Species Sequence Comparison Leila Taher1, Oliver Rinner2,3, Saurabh Garg1, Alexander Sczyrba4 and Burkhard Morgenstern5,*
Nucleic Acids Research, 2004, Vol. 32, Web Server issue W305–W308 DOI: 10.1093/nar/gkh386 AGenDA: gene prediction by cross-species sequence comparison Leila Taher1, Oliver Rinner2,3, Saurabh Garg1, Alexander Sczyrba4 and Burkhard Morgenstern5,* 1International Graduate School for Bioinformatics and Genome Research, University of Bielefeld, Postfach 10 01 31, 33501 Bielefeld, Germany, 2GSF Research Center, MIPS/Institute of Bioinformatics, Ingolsta¨dter Landstrasse 1, 85764 Neuherberg, Germany, 3Brain Research Institute, ETH Zuurich,€ Winterthurerstrasse 190, CH-8057 Zuurich,€ Switzerland, 4Faculty of Technology, Research Group in Practical Computer Science, University of Bielefeld, Postfach 10 01 31, 33501 Bielefeld, Germany and 5Institute of Microbiology and Genetics, University of Go¨ttingen, Goldschmidtstrasse 1, 37077 Go¨ttingen, Germany Received March 5, 2004; Revised and Accepted March 24, 2004 ABSTRACT Thus, with the huge amount of data produced by sequencing projects, the development of high-quality gene-prediction Automatic gene prediction is one of the major tools has become a priority in bioinformatics. For eukaryotic challenges in computational sequence analysis. organisms, gene prediction is particularly challenging, since Traditional approaches to gene finding rely on statis- protein-coding exons are usually separated by introns of vari- tical models derived from previously known genes. By able length. Most traditional methods for gene prediction are contrast, a new class of comparative methods relies intrinsic methods; they use hidden Markov models (HMMs) or on comparing genomic sequences from evolutionary other stochastic approaches describing the statistical composi- related organisms to each other. These methods are tion of introns, exons, etc. Such models are trained with already based on the concept of phylogenetic footprinting: known genes from the same or a closely related species; the they exploit the fact that functionally important most popular of these tools is GenScan (1). -

Mammal Survey Pol'ana (Slovakia) 2005 5�
MAMMAL SURVEY POL 'ANA (S LOVAKIA ) 2005 RESULTS OF TEN DAYS ’ FIELD WORK RAPPORT 2009.23 Juli 2009 Uitgave van de Veldwerkgroep van de Zoogdiervereniging MAMMAL SURVEY POL 'ANA (S LOVAKIA ) 2005 RESULTS OF TEN DAYS ’ FIELD WORK Editors Jan Buys Jeroen Willemsen Translations Karin de Bie Authors Jan Boshamer Jan Buys Annemarie van Diepenbeek Rob Koelman Jeroen van der Kooij Rudy van der Kuil Kees Mostert Janny Resoort Froukje Rienks Kamiel Spoelstra Anke van der Wal Jeroen Willemsen Illustration on front cover Froukje Rienks Photographs Jan Boshamer Jan Buys Jeroen van der Kooij Rudy van der Kuil Janny Resoort Kamiel Spoelstra Joost Verbeek Anke van der Wal Uitgave van de Veldwerkgroep van de Zoogdiervereniging Rapport 2009.23 Arnhem, juli 2009 ISBN: 978-90-79924-12-7 All publications of the Veldwerkgroep can be downloaded free of charge from our website: http://www.zoogdiervereniging.nl/node/137 The English page of the website contains all publications in English: http://www.zoogdiervereniging.nl/node/411 SUMMARY by: Jan Buys and Jeroen Willemsen From July 27th until August 4th 2005, the Veldwerkgroep (VWG) of the Dutch Mammal Society (Zoogdiervereniging) paid a visit to the Pol’ana Biosphere reserve in Slovakia. A workshop in the summer is a traditional part of the VWG’s yearly program, aimed at surveying mammal species which are rare or absent in the Netherlands, and to extend and exchange knowledge of survey methods and species. Secondly, these workshops abroad aim to enlarge the knowledge of presence and abundance of mammals, and to a lesser extent of other fauna groups, in the area visited. -

Revised and Commented Checklist of Mammal Species of the Romanian Fauna
REVISED AND COMMENTED CHECKLIST OF MAMMAL SPECIES OF THE ROMANIAN FAUNA DUMITRU MURARIU Abstract. Due to the permanent influences of different factors (habitat degradation andfragmentation, deforestation, infrastructure and urbanization, natural extension or decreasing of some species’ distribution, increasing number of alien species etc.), from time to time the faunistic structure of a certain area is changing. As a result of the permanent and increasing anthropic and invasive species’ pressure, our previous checklist of recent mammals from Romania (since 1984) became out of date. A number of 108 taxa are mentioned in this checklist, representing 7 orders of mammals: Insectivora (10 species), Chiroptera (30 sp.), Lagomorpha (2 sp.), Rodentia (35 sp.), Cetacea (3 sp.), Carnivora (19 sp.), Artiodactyla (8 sp.). In this list are mentioned the scientific and vernacular names (in Romanian and English languages), species distribution and conservation status, according to the Romanian regulations. Thus, only 21 species have stable populations while 76 have populations in decline or in drastic decline. Other categories are not evaluated or even present an increase in their population. Key words: species and subspecies, recent mammals, distribution, conservation. 1. INTRODUCTION MURARIU published ‘La Liste de Mammifère actuels de Roumanie; noms scientifiques et Roumains’ in 1984. That moment represented an astonishing progress in the knowledge of mammals, only in two decades. A number of 3,500 species (belonging to 1,000 genera) were reported by DAVID & GOLLEY (1965) and 4,170 species were reported by HONACKI et al. (1982). Description of new mammal species is continuing, and WILSON & REEDER DEEANN (2005) presented “... the taxonomic classification and distribution of the more than 5,400 species of mammals that exist today”. -

Seasonal Changes in Tawny Owl (Strix Aluco) Diet in an Oak Forest in Eastern Ukraine
Turkish Journal of Zoology Turk J Zool (2017) 41: 130-137 http://journals.tubitak.gov.tr/zoology/ © TÜBİTAK Research Article doi:10.3906/zoo-1509-43 Seasonal changes in Tawny Owl (Strix aluco) diet in an oak forest in Eastern Ukraine 1, 2 Yehor YATSIUK *, Yuliya FILATOVA 1 National Park “Gomilshanski Lisy”, Kharkiv region, Ukraine 2 Department of Zoology and Animal Ecology, Faculty of Biology, V.N. Karazin Kharkiv National University, Kharkiv, Ukraine Received: 22.09.2015 Accepted/Published Online: 25.04.2016 Final Version: 25.01.2017 Abstract: We analyzed seasonal changes in Tawny Owl (Strix aluco) diet in a broadleaved forest in Eastern Ukraine over 6 years (2007– 2012). Annual seasons were divided as follows: December–mid-April, April–June, July–early October, and late October–November. In total, 1648 pellets were analyzed. The most important prey was the bank vole (Myodes glareolus) (41.9%), but the yellow-necked mouse (Apodemus flavicollis) (17.8%) dominated in some seasons. According to trapping results, the bank vole was the most abundant rodent species in the study region. The most diverse diet was in late spring and early summer. Small forest mammals constituted the dominant group in all seasons, but in spring and summer their share fell due to the inclusion of birds and the common spadefoot (Pelobates fuscus). Diet was similar in late autumn, before the establishment of snow cover, and in winter. The relative representation of species associated with open spaces increased in winter, especially in years with deep snow cover, which may indicate seasonal changes in the hunting habitats of the Tawny Owl. -

A New Efficient Method to Detect Genetic Interactions for Lung Cancer
Luyapan et al. BMC Med Genomics (2020) 13:162 https://doi.org/10.1186/s12920-020-00807-9 TECHNICAL ADVANCE Open Access A new efcient method to detect genetic interactions for lung cancer GWAS Jennifer Luyapan1,2, Xuemei Ji2, Siting Li1,2, Xiangjun Xiao3, Dakai Zhu2,3, Eric J. Duell4, David C. Christiani5,6, Matthew B. Schabath7, Susanne M. Arnold8, Shanbeh Zienolddiny9, Hans Brunnström10, Olle Melander11, Mark D. Thornquist12, Todd A. MacKenzie1,2, Christopher I. Amos1,2,3* and Jiang Gui1,2* Abstract Background: Genome-wide association studies (GWAS) have proven successful in predicting genetic risk of disease using single-locus models; however, identifying single nucleotide polymorphism (SNP) interactions at the genome- wide scale is limited due to computational and statistical challenges. We addressed the computational burden encountered when detecting SNP interactions for survival analysis, such as age of disease-onset. To confront this problem, we developed a novel algorithm, called the Efcient Survival Multifactor Dimensionality Reduction (ES-MDR) method, which used Martingale Residuals as the outcome parameter to estimate survival outcomes, and imple- mented the Quantitative Multifactor Dimensionality Reduction method to identify signifcant interactions associated with age of disease-onset. Methods: To demonstrate efcacy, we evaluated this method on two simulation data sets to estimate the type I error rate and power. Simulations showed that ES-MDR identifed interactions using less computational workload and allowed for adjustment of covariates. We applied ES-MDR on the OncoArray-TRICL Consortium data with 14,935 cases and 12,787 controls for lung cancer (SNPs 108,254) to search over all two-way interactions to identify genetic interactions associated with lung cancer age-of-onset.= We tested the best model in an independent data set from the OncoArray-TRICL data.