CICUAL; Approval Number C.FUA 14-023
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Repositiorio | FAUBA | Artículos De Docentes E Investigadores De FAUBA
Biodivers Conserv (2011) 20:3077–3100 DOI 10.1007/s10531-011-0118-9 REVIEW PAPER Effects of agriculture expansion and intensification on the vertebrate and invertebrate diversity in the Pampas of Argentina Diego Medan • Juan Pablo Torretta • Karina Hodara • Elba B. de la Fuente • Norberto H. Montaldo Received: 23 July 2010 / Accepted: 15 July 2011 / Published online: 24 July 2011 Ó Springer Science+Business Media B.V. 2011 Abstract In this paper we summarize for the first time the effects of agriculture expansion and intensification on animal diversity in the Pampas of Argentina and discuss research needs for biodiversity conservation in the area. The Pampas experienced little human intervention until the last decades of the 19th century. Agriculture expanded quickly during the 20th century, transforming grasslands into cropland and pasture lands and converting the landscape into a mosaic of natural fragments, agricultural fields, and linear habitats. In the 1980s, agriculture intensification and replacement of cattle grazing- cropping systems by continuous cropping promoted a renewed homogenisation of the most productive areas. Birds and carnivores were more strongly affected than rodents and insects, but responses varied within groups: (a) the geographic ranges and/or abundances of many native species were reduced, including those of carnivores, herbivores, and specialist species (grassland-adapted birds and rodents, and probably specialized pollinators), sometimes leading to regional extinction (birds and large carnivores), (b) other native species were unaffected (birds) or benefited (bird, rodent and possibly generalist pollinator and crop-associated insect species), (c) novel species were introduced, thus increasing species richness of most groups (26% of non-rodent mammals, 11.1% of rodents, 6.2% of birds, 0.8% of pollinators). -
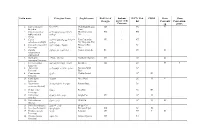
Red List of Endemic IUCN Red CITES Bern Bonn Georgia Species of the List Conventi Convention Caucasus on (CMS) 1
Latin name Georgian Name English name Red List of Endemic IUCN Red CITES Bern Bonn Georgia species of the list Conventi Convention Caucasus on (CMS) 1. Capra aegagrus niamori Wild Goat, Bezoar CR VU II Erxleben. Goat 2. Capra caucasica dasavleTkavkasiuri West Caucasian EN + EN Güldenstädt & jixvi Tur Pallas. 3. Capra aRmosavleTkavkasiuri East Caucasian VU + NT cylindricornis Blyth. jixvi Tur, Dagestan Tur 4. Capreolus capreolus evropuli Sveli European Roe LC Linnaeus. Deer 5. Gazella qurciki, jeirani Goitered Gazelle RE VU II subgutturosa Güldenstädt. 6. Rupicapra arCvi, fsiti Northern Chamois EN LC II rupicapra Linnaeus. 7. Cervus elaphus keTilSobili iremi Red Deer CR LC II I Linnaeus. 8. Sus scrofa gareuli Rori, taxi Eurasian Wild LC Linnaeus. Boar 9. Canis aureus tura Golden Jackal LC III Linnaeus. 10. Canis lupus mgeli Grey Wolf LC II II Linnaeus. 11. Nyctereutes enotisebri ZaRli Racoon Dog LC procyonoides Gray. 12. Vulpes vulpes mela Red Fox LC III Linnaeus. 13. Felis chaus lelianis kata Jungle Cat VU LC II Schreber. 14. Felis silvestris tyis kata Wild Cat LC II II Shreber. 15. Felis libyca Forster. velis kata Steppe Cat 16. Lynx lynx Linnaeus. focxveri Eurasian Lynx CR LC II 17. Panthera pardus jiqi Leopard CR NT I II Linnaeus. 18. Hyaena hyaena afTari Striped Hyaena CR NT Linnaeus. 19. Lutra lutra wavi Eurasian Otter, VU NT I II Linnaeus. Common Otter 20. Martes foina kldis kverna Stone Marten, LC III Erxleben. Beech Marten 21. Martes martes tyis kverna European Pine LC Linnaeus. Marten 22. Meles meles maCvi Eurasian Badger LC Linnaeus. 23. Mustela lutreola waula European Mink EN II Linnaeus. -

Ecology and Status of the Jaguarundi Puma Yagouaroundi: a Synthesis of Existing Knowledge
Giordano, A. J. (2015). Ecology and status of the jaguarundi Puma yagouaroundi: a synthesis of existing knowledge. Mammal Review 46: 30-43. Keywords: 2MX/diet/distribution/ecology/food/habitat/Herpailurus yagouaroundi/home range/home range size/human-wildlife conflict/jaguarundi/movement/prey/prey species/Puma yagouaroundi/review/status/threats Abstract: 1. The ecology of the jaguarundi is poorly known, so I reviewed the literature for all original data and remarks on jaguarundi observations, ecology, and behaviour, to synthesize what is known about the species. 2. Jaguarundis occupy and use a range of habitats with dense undergrowth from northern Mexico to central Argentina, but may be most abundant in seasonal dry, Atlantic, gallery, and mixed grassland/agricultural forest landscapes. 3. Jaguarundis are principally predators of small (sigmodontine) rodents, although other mammals, birds, and squamate reptiles are taken regularly. 4. The vast majority of jaguarundi camera-trap records occurred during daylight hours (0600 h-1800 h); jaguaurndis are also predominantly terrestrial, although they appear to be capable tree climbers. 5. Home range sizes for jaguarundis vary greatly, but most are .25 km2; females' territories may be much smaller than or similar in size to those of males. Males may concentrate movements in one area before shifting to another and, as with other felids, intersexual overlap in habitat use appears to be common. 6. Interference competition may be important in influencing the distribution and ecology of jaguarundis, although their diurnal habits may somewhat mitigate its effect. 7. Conflict between humans and jaguarundis over small livestock may be widespread among rural human communities and is likely to be underreported. -

Mammal Survey Pol'ana (Slovakia) 2005 5�
MAMMAL SURVEY POL 'ANA (S LOVAKIA ) 2005 RESULTS OF TEN DAYS ’ FIELD WORK RAPPORT 2009.23 Juli 2009 Uitgave van de Veldwerkgroep van de Zoogdiervereniging MAMMAL SURVEY POL 'ANA (S LOVAKIA ) 2005 RESULTS OF TEN DAYS ’ FIELD WORK Editors Jan Buys Jeroen Willemsen Translations Karin de Bie Authors Jan Boshamer Jan Buys Annemarie van Diepenbeek Rob Koelman Jeroen van der Kooij Rudy van der Kuil Kees Mostert Janny Resoort Froukje Rienks Kamiel Spoelstra Anke van der Wal Jeroen Willemsen Illustration on front cover Froukje Rienks Photographs Jan Boshamer Jan Buys Jeroen van der Kooij Rudy van der Kuil Janny Resoort Kamiel Spoelstra Joost Verbeek Anke van der Wal Uitgave van de Veldwerkgroep van de Zoogdiervereniging Rapport 2009.23 Arnhem, juli 2009 ISBN: 978-90-79924-12-7 All publications of the Veldwerkgroep can be downloaded free of charge from our website: http://www.zoogdiervereniging.nl/node/137 The English page of the website contains all publications in English: http://www.zoogdiervereniging.nl/node/411 SUMMARY by: Jan Buys and Jeroen Willemsen From July 27th until August 4th 2005, the Veldwerkgroep (VWG) of the Dutch Mammal Society (Zoogdiervereniging) paid a visit to the Pol’ana Biosphere reserve in Slovakia. A workshop in the summer is a traditional part of the VWG’s yearly program, aimed at surveying mammal species which are rare or absent in the Netherlands, and to extend and exchange knowledge of survey methods and species. Secondly, these workshops abroad aim to enlarge the knowledge of presence and abundance of mammals, and to a lesser extent of other fauna groups, in the area visited. -

Revised and Commented Checklist of Mammal Species of the Romanian Fauna
REVISED AND COMMENTED CHECKLIST OF MAMMAL SPECIES OF THE ROMANIAN FAUNA DUMITRU MURARIU Abstract. Due to the permanent influences of different factors (habitat degradation andfragmentation, deforestation, infrastructure and urbanization, natural extension or decreasing of some species’ distribution, increasing number of alien species etc.), from time to time the faunistic structure of a certain area is changing. As a result of the permanent and increasing anthropic and invasive species’ pressure, our previous checklist of recent mammals from Romania (since 1984) became out of date. A number of 108 taxa are mentioned in this checklist, representing 7 orders of mammals: Insectivora (10 species), Chiroptera (30 sp.), Lagomorpha (2 sp.), Rodentia (35 sp.), Cetacea (3 sp.), Carnivora (19 sp.), Artiodactyla (8 sp.). In this list are mentioned the scientific and vernacular names (in Romanian and English languages), species distribution and conservation status, according to the Romanian regulations. Thus, only 21 species have stable populations while 76 have populations in decline or in drastic decline. Other categories are not evaluated or even present an increase in their population. Key words: species and subspecies, recent mammals, distribution, conservation. 1. INTRODUCTION MURARIU published ‘La Liste de Mammifère actuels de Roumanie; noms scientifiques et Roumains’ in 1984. That moment represented an astonishing progress in the knowledge of mammals, only in two decades. A number of 3,500 species (belonging to 1,000 genera) were reported by DAVID & GOLLEY (1965) and 4,170 species were reported by HONACKI et al. (1982). Description of new mammal species is continuing, and WILSON & REEDER DEEANN (2005) presented “... the taxonomic classification and distribution of the more than 5,400 species of mammals that exist today”. -

Seasonal Changes in Tawny Owl (Strix Aluco) Diet in an Oak Forest in Eastern Ukraine
Turkish Journal of Zoology Turk J Zool (2017) 41: 130-137 http://journals.tubitak.gov.tr/zoology/ © TÜBİTAK Research Article doi:10.3906/zoo-1509-43 Seasonal changes in Tawny Owl (Strix aluco) diet in an oak forest in Eastern Ukraine 1, 2 Yehor YATSIUK *, Yuliya FILATOVA 1 National Park “Gomilshanski Lisy”, Kharkiv region, Ukraine 2 Department of Zoology and Animal Ecology, Faculty of Biology, V.N. Karazin Kharkiv National University, Kharkiv, Ukraine Received: 22.09.2015 Accepted/Published Online: 25.04.2016 Final Version: 25.01.2017 Abstract: We analyzed seasonal changes in Tawny Owl (Strix aluco) diet in a broadleaved forest in Eastern Ukraine over 6 years (2007– 2012). Annual seasons were divided as follows: December–mid-April, April–June, July–early October, and late October–November. In total, 1648 pellets were analyzed. The most important prey was the bank vole (Myodes glareolus) (41.9%), but the yellow-necked mouse (Apodemus flavicollis) (17.8%) dominated in some seasons. According to trapping results, the bank vole was the most abundant rodent species in the study region. The most diverse diet was in late spring and early summer. Small forest mammals constituted the dominant group in all seasons, but in spring and summer their share fell due to the inclusion of birds and the common spadefoot (Pelobates fuscus). Diet was similar in late autumn, before the establishment of snow cover, and in winter. The relative representation of species associated with open spaces increased in winter, especially in years with deep snow cover, which may indicate seasonal changes in the hunting habitats of the Tawny Owl. -

Pdf (Last Access Plínio Barbosa De Camargo: Contribution to Data Analysis and on 03/Apr/2017)
Biota Neotropica 18(2): e20170395, 2018 www.scielo.br/bn ISSN 1676-0611 (online edition) Inventory Human-modified landscape acts as refuge for mammals in Atlantic Forest Alex Augusto de Abreu Bovo1 , Marcelo Magioli1 , Alexandre Reis Percequillo1 , Cecilia Kruszynski2,3 , Vinicius Alberici1 , Marco A. R. Mello4 , Lidiani Silva Correa1, João Carlos Zecchini Gebin1, Yuri Geraldo Gomes Ribeiro1 , Francisco Borges Costa5, Vanessa Nascimento Ramos5, Hector Ribeiro Benatti5, Beatriz Lopes1, Maísa Z. A. Martins1, Thais Rovere Diniz-Reis2 , Plínio Barbosa de Camargo6, Marcelo Bahia Labruna5 & Katia Maria Paschoaletto Micchi de Barros Ferraz1* 1Universidade de São Paulo, Escola Superior de Agricultura “Luiz de Queiroz”, Departamento de Ciências Florestais, Laboratório de Ecologia, Manejo e Conservação da Fauna Silvestre, Av. Pádua Dias, 11, 13418-900, Piracicaba, SP, Brasil 2Universidade de São Paulo, Centro de Energia Nuclear na Agricultura, Piracicaba, SP, Brasil 3Leibniz Institut fur Zoo und Wildtierforschung eV, Berlin, Germany 4Universidade Federal de Minas Gerais, Biologia Geral, Belo Horizonte, MG, Brasil 5Universidade de São Paulo, Faculdade de Medicina Veterinária e Zootecnia, Departamento de Medicina Veterinária Preventiva e Saúde Animal, São Paulo, SP Brasil 6Universidade de São Paulo, Centro de Energia Nuclear na Agricultura, CENA - Laboratório de Ecologia Isotópica, Piracicaba, SP, Brasil *Corresponding author: Katia Maria Paschoaletto Micchi de Barros Ferraz, e-mail: [email protected] BOVO, A.A.A, MAGIOLI, M.; PERCEQUILLO, A.R., KRUSZYNSKI, C., ALBERICI, V., MELLO, M.A.R., CORREA, L.S., GEBIN, J.C.Z., RIBEIRO, Y.G.G., COSTA, F.B.; RAMOS, V.N., BENATTI, H.R., LOPES, B., MARTINS, M.Z.A., DINIZ-REIS, T.R., CAMARGO, P.B.; LABRUNA, M.B., FERRAZ, K.M.P.M.B. -

A Review of the Results Obtained During the Field Study Group Summer Camps of the Dutch Mammal Society, 1986-2014
A review of the results obtained during the Field Study Group summer camps of the Dutch Mammal Society, 1986-2014 Jan Piet Bekker1, Kees Mostert2, Jan P.C. Boshamer3 & Eric Thomassen4 1Zwanenlaan 10, NL-4351 RX Veere, the Netherlands, e-mail: [email protected] 2Palamedesstraat 74, NL-2612 XS Delft, the Netherlands 3Vogelzand 4250, NL-1788 MP Den Helder, the Netherlands 4Middelstegracht 28a, NL-2312 TX Leiden, the Netherlands Abstract: The 28 summer camps of the Field Study Group of the Dutch Mammal Society organised between 1986 and 2014 are reviewed here. Over time the Field Study Group gradually spread out its activities throughout Europe, including former Eastern Bloc countries. Camp locations were found through contacts in host countries, who also assist in the preparation of camp activities. Out of a total of 160 participants from the Netherlands and Belgium, 80 attended a summer camp once and 80 joined more than once; 116 participants from local origin were active dur- ing these camps. For the 128 mammal species found, the observation techniques used are described. Overall, 7,662 small mammals were caught with live-traps and 990 bats were caught in mist nets. Among the trapped mammals, 421 casualties were counted, predominantly common and pygmy shrews in northern European countries. In pellets, pre- dominantly from barn owls, 21,620 small mammals were found. With detectors, 3,908 bats could be identified. Caves and (old) buildings were explored for bats, and the results of these surveys made up a large part of the total number of bats found. Sightings (> 1,740) and tracks & signs (> 1,194) revealed most of all the presence of carnivora and even- toed ungulates (Artiodactyla). -

Molecular Detection of Leishmania Spp. in Road-Killed Wild Mammals In
Richini-Pereira et al. Journal of Venomous Animals and Toxins including Tropical Diseases 2014, 20:27 http://www.jvat.org/content/20/1/27 RESEARCH Open Access Molecular detection of Leishmania spp. in road-killed wild mammals in the Central Western area of the State of São Paulo, Brazil Virginia Bodelão Richini-Pereira, Pamela Merlo Marson, Enio Yoshinori Hayasaka, Cassiano Victoria, Rodrigo Costa da Silva and Hélio Langoni* Abstract Background: Road-killed wild animals have been classified as sentinels for detecting such zoonotic pathogens as Leishmania spp., offering new opportunities for epidemiological studies of this infection. Methods: This study aimed to evaluate the presence of Leishmania spp. and Leishmania chagasi DNA by PCR in tissue samples (lung, liver, spleen, kidney, heart, mesenteric lymph node and adrenal gland) from 70 road-killed wild animals. Results: DNA was detected in tissues of one Cavia aperea (Brazilian guinea pig), five Cerdocyon thous (crab-eating fox), one Dasypus septemcinctus (seven-banded armadillo), two Didelphis albiventris (white-eared opossum), one Hydrochoerus hydrochoeris (capybara), two Myrmecophaga tridactyla (giant anteater), one Procyon cancrivorus (crab-eating raccoon), two Sphiggurus spinosus (porcupine) and one Tamandua tetradactyla (lesser anteater) from different locations in the Central Western part of São Paulo state. The Leishmania chagasi DNA were confirmed in mesenteric lymph node of one Cerdocyon thous. Results indicated common infection in wild animals. Conclusions: The approach employed -

Food Supply (Orthoptera, Mantodea, Rodentia and Eulipotyphla)
Slovak Raptor Journal 2017, 11: 1–14. DOI: 10.1515/srj-2017-0005. © Raptor Protection ofSlovakia (RPS) Food supply (Orthoptera, Mantodea, Rodentia and Eulipotyphla) and food preferences of the red-footed falcon (Falco vespertinus) in Slovakia Potravná ponuka (Orthoptera, Mantodea, Rodentia a Eulipotyphla) a potravné preferencie sokola kobcovitého (Falco vespertinus) na Slovensku Anton KRIŠTÍN, Filip TULIS, Peter KLIMANT, Kristián BACSA & Michal AMBROS Abstract: Food supply in the nesting territories of species has a key role to the species diet composition and their breeding success. Red-footed falcon (Falco vespertinus) preys predominantly on larger insect species with a supplementary portion of smaller vertebrates. In the breeding periods 2014 and 2016 their food supply, focusing on Orthoptera, Mantodea, Rodentia and Eulipotyphla, was analysed at five historical nesting sites of the species in Slovakia. Preference for these prey groups in the diet was also studied at the last active nesting site in this country. Overall we recorded 45 Orthoptera species (of which 23 species are known as the food of the red-footed falcon), one species of Mantodea, 10 species of Rodentia (of which 2 species are known as the food of the red-footed falcon) and 5 species of the Eulipotyphla order in the food supply. With regard to the availability of the falcons' preferred food, in both years the most suitable was the Tvrdošovce site, which continuously showed the greatest range and abundance of particular species. In the interannual comparison the insects showed lower variability in abundance than the small mammals. In 2014 the growth of the common vole (Microtus arvalis) population culminated and with the exception of a single site (Bodza) a slump in abundance was recorded in 2016. -

Animals of the Cloud Forest: Isotopic Variation of Archaeological Faunal Remains from Kuelap, Peru
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2018 Animals of the Cloud Forest: Isotopic Variation of Archaeological Faunal Remains from Kuelap, Peru Samantha Michell University of Central Florida Part of the Anthropology Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Masters Thesis (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Michell, Samantha, "Animals of the Cloud Forest: Isotopic Variation of Archaeological Faunal Remains from Kuelap, Peru" (2018). Electronic Theses and Dissertations, 2004-2019. 5942. https://stars.library.ucf.edu/etd/5942 ANIMALS OF THE CLOUD FOREST: ISOTOPIC VARIATION OF ARCHAEOLOGICAL FAUNAL REMAINS FROM KUELAP, PERU by SAMANTHA MARIE MICHELL B.S. Idaho State University, 2014 A thesis submitted in partial fulfillment of the requirements for the degree of Master of Arts in the Department of Anthropology in the College of Sciences at the University of Central Florida Orlando, Florida Summer Term 2018 © 2018 Samantha Michell ii ABSTRACT Stable isotopic analyses of faunal remains are used as a proxy for reconstructing the ancient Chachapoya dietary environment of the northeastern highlands in Peru. Archaeologists have excavated animal remains from refuse piles at the monumental center of Kuelap (AD 900-1535). This archaeological site is located at 3000 meters above sea level (m.a.s.l.), where C3 plants dominate the region. -

An Ecological and Conserv an Ecolog
Mammals of the Bodoquena Mountains, southwestern Brazil: an ecological and conservation analysis Nilton C. Cáceres 1; Marcos R. Bornschein 2; Wellington H. Lopes 1 & Alexandre R. Percequillo 3 1 Departamento de Biologia, CCNE, Universidade Federal de Santa Maria. Faixa de Camobi, km 9, 97110-970 Santa Maria, Rio Grande do Sul, Brasil. 2 Liga Ambiental. Rua Olga de Araújo Espíndola 1380, bloco N, ap. 31, 81050-280 Curitiba, Paraná, Brasil. 3 Departamento de Ciências Biológicas, Escola Superior de Agricultura “Luiz de Queiroz”, Universidade de São Paulo. Avenida Pádua Dias 11, Caixa Postal 9, 13418-900 Piracicaba, São Paulo, Brasil. ABSTRACT. We carried out a mammalian survey in the neighborhoods of the Serra da Bodoquena National Park, Mato Grosso do Sul state, a region poorly known in southwestern Brazil. During the months of April, May and July 2002 we used wire live trap, direct observation, indirect evidence (e.g. tracks), carcasses, and interviews with local residents to record mammalian species. Fifty six mammal species were recorded, including threatened species (14%). These records were discussed regarding species abundance, distribution, range extension, habitat, and conservation. The geographic distribution and ecology of the poorly known marsupials Thylamys macrurus and Micoureus constantiae in Brazil are emphasized. KEY WORDS. Brazilian savanna; deciduous forest; distribution; Mammalia; species richness. RESUMO. Mamíferos da Serra da BodoquenaBodoquena, sudoeste do Brasilasil: uma análise ecológica e conservacionista. Efetuamos um levantamento de espécies de mamíferos no entorno do Parque Nacional da Serra da Bodoquena, Estado de Mato Grosso do Sul, uma região pouco conhecida no sudoeste do Brasil. Durante os meses de abril, maio e julho de 2002 efetuamos captura com armadilhas, observação direta, busca por evidências indiretas (e.g.