Measurement of Transference Numbers of the Bisulfate Ion in Aqueous Sulfuric Acid
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

4월 24일(목), 09:00~10:30
포스터 I: 4월 24일(목), 09:00~10:30 공업화학: 4월 24일(목), 09:00 ~ 10:30 좌장: 박정훈(동국대) Cleaning system study with mounting activated species of solution using P공업목-1(118) (강원대)권흥수, 이원규 atmospheric pressure plasma 기공구조 조절 제올라이트 촉매를 이용한 액체연료의 흡열량 향상 (고려대)현동훈, 김중연, 김유리, 전병희, P공업목-2(118) (우수 포스터 발표상 후보) 김성현, (국방과학(연))정병훈, 한정식 (고려대)신동건, 김성현, 이창훈, P공업목-3(118) 열 안정성이 우수한 CO 흡착제용 다공성 유무기 복합 지지체 제조 2 조동현, 정현철 (고려대)김유리, 김성현, 전병희, P공업목-4(119) 분산제의 구조에 따른 Jet A-1 연료의 열산화반응 탄소침적물 감소 연구 김중연, 현동훈 (국방과학(연))한정식, 정병훈 Effect of SAA Pretreatment on Temperature of Enzymatic Hydrolysis Using P공업목-5(119) (경기대)장서윤, 박장한, 김준석 Lignocellulosic Biomass Pretreatment and fractionation of Helianthus tuberosus residue by flow-through P공업목-6(119) (경기대)박용철, 박장한, 김준석 reaction (순천대)오성준 (순천대)양지선, 박권필 P공업목-7(119) Carbon Felt 산처리에 의한 바나듐 레독스 흐름전지에 대한 영향 ((주)CNL Energy)나일채, 이정훈 ((주)ETIS)추천호 메틸카바메이트 및 메탄올을 이용한 디메틸카보네이트 제조를 위한 고활성 P공업목-8(120) (한양대)이재홍, 서영웅 촉매 조사 (우수 포스터 발표상 후보) 연속흐름반응기에서 고농도 NaBH 가수분해 반응의 촉매 종류에 따른 P공업목-9(120) 4 (순천대)오성준, 정현승, 박권필 수소발생연구 (순천대)안명원, 곽동영, 복영빈, 박권필 P공업목-10(120) 해조류 종류별 후코산틴 추출 및 염분세척 ((주) ETIS)추천호, 김영숙 (순천대)이세훈, 박권필, ((주)ETIS)김영숙, P공업목-11(120) 효소연료전지에서 Anode 조건에 따른 OCV 변화 추천호, ((주)CNL Energy)나일채 (순천대)정재현, 정회범, 박권필 P공업목-12(121) PEMFC MEA제법에 의한 성능 및 내구성 향상 ((주)CNL Energy)라일채, 이 호 (순천대)정재현, 신은경, 정회범, 박권필 P공업목-13(121) PEM 수전해 과정에서 전극과 전해질 막의 변화 ((주)CNL Energy)라일채, 이 호 (순천대)정재현, 박권필 P공업목-14(121) 직접 개미산 연료전지(DFAFC)의 구동 조건에 따른 성능 비교 ((주)CNL Energy)나일채 (순천대)정재진, 박권필 P공업목-15(121) 가속화도 4.0의 전극, 막, MEA 가속화 기법 개발 (현대자동차)안병기, 김세훈, 고재준 (순천대)정재진, 박권필 P공업목-16(122) 탄화수소막을 사용한 -

Preparing to Manufacture Hydrogen Peroxide
PREPARING TO MANUFACTURE HYDROGEN PEROXIDE Part of the Hydrogen Peroxide Propulsion Guide The early laboratory preparation of hydrogen peroxide was based on the technique that Thenard used during the initial preparation of hydrogen peroxide. In this technique, barium nitrate, purified by recrystallization, was decomposed by heating in air in a porcelain retort. The resulting oxide was further oxidized by heating in a stream of oxygen to a dull red heat. The barium peroxide which formed was then dampened, ground, and dissolved in hydrochloric acid (nitric acid was used in Thenard’s initial experiments). A slight excess of sulfuric acid was then added to precipitate barium sulfate and regenerate hydrochloric acid. The procedure of barium peroxide solution and sulfate precipitation was repeated several times in the same solution to increase the peroxide concentration (concentrations of up to 33 percent by weight hydrogen peroxide could be achieved in this manner). The concentrated solution containing water, hydrogen peroxide, and hydrochloric acid, along with accumulated impurities, was cooled with ice and saturated with barium peroxide; iron and manganese impurities in the solution were then precipitated out as phosphates. The hydrochloric acid was removed by the addition of silver sulfate and the sulfate ion was removed by the subsequent addition of barium oxide. Further concentration was accomplished by vacuum distillation until “no further density increase occurs.” Thenard reported that 100 w/o hydrogen peroxide (on the basis of density data and the measurement of the volume of oxygen released) could be obtained by this technique. The first record of commercial production of hydrogen peroxide appeared in the 1865 to 1875 period. -

Most Common Substances in Chemicals for Different Industries
Table 14 Most common substances by industry Figures for 2009. Substances contained in more than 10 products. A substance can be omitted for secrecy reasons. Industry CAS-no Substance Number of products A01 Agriculture 7732-18-5 Water 458 6484-52-2 Nitric acid ammonium salt 77 57-55-6 1,2-Propanediol 75 64742-65-0 Distillates (petroleum), solvent-dewaxed heavy 74 paraffinic 63148-62-9 Siloxanes and Silicones, di-Me 60 57-13-6 Urea 54 67-63-0 2-Propanol 54 11138-66-2 Xanthan gum 53 7783-20-2 Sulfuric acid diammonium salt 53 7722-76-1 Phosphoric acid, monoammonium salt 52 7487-88-9 Sulfuric acid magnesium salt (1:1) 51 7447-40-7 Potassium chloride 46 56-81-5 1,2,3-Propanetriol 44 7778-80-5 Sulfuric acid dipotassium salt 43 1310-73-2 Sodium hydroxide 41 7647-14-5 Sodium chloride 41 2634-33-5 1,2-Benzisothiazol-3(2H)-one 38 1310-58-3 Potassium hydroxide 33 7757-79-1 Nitric acid potassium salt 33 7785-87-7 Manganese sulfate 32 7664-38-2 Phosphoric acid 32 72623-87-1 Lubricating oils, petroleum, C20-50, 31 hydrotreated neutral oil-based 64741-89-5 Distillates (petroleum), solvent-refined light 29 paraffinic 77-92-9 1,2,3-Propanetricarboxylic acid, 2-hydroxy- 28 8061-51-6 Lignosulfonic acid, sodium salt 28 7778-18-9 Sulfuric acid, calcium salt (1:1) 27 64-17-5 Ethanol 27 14807-96-6 Talc 26 64742-94-5 Solvent naphtha (petroleum), heavy arom. 25 68649-42-3 Phosphorodithioic acid, O,O-di-C1-14-alkyl 24 esters, zinc salts (2:1) 64-02-8 Glycine, N,N'-1,2-ethanediylbis[N- 24 (carboxymethyl)-, tetrasodium salt 64-19-7 Acetic acid 23 99-76-3 Benzoic acid, -
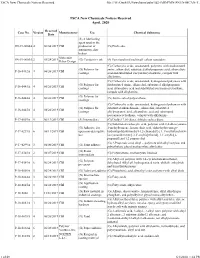
TSCA New Chemicals Notices Received File:///W:/Oneepa/Newchems/Pubs/5D2-IMD/PMN-SNUN-MCAN-T
TSCA New Chemicals Notices Received file:///W:/OneEPA/Newchems/pubs/5d2-IMD/PMN-SNUN-MCAN-T... TSCA New Chemicals Notices Received April, 2020 Received Case No. Version Manufacturer Use Chemical Substance Date (S) A lubricating agent used in the SN-19-0004A 4 06/04/2019 CBI production of (G) Pitch coke automotive disc brakes Molecular SN-19-0005A 2 05/28/2019 (G) Conductive ink (S) Functionalized multiwall carbon nanotubes Rebar Design (G) Carboxylic acids, unsaturated, polymers with disubstituted (G) Polymer for amine, alkanediol, substituted alkylpropanoic acid, alkanedioic P-16-0442A 4 06/26/2019 CBI coatings acid and substituted isocyanatocycloalkane, compds with alkylamine (G) Carboxylic acids, unsaturated, hydrogenated polymers with (G) Polymer for disubstituted amine, alkanediol, substituted alkylpropanoic P-16-0443A 4 06/26/2019 CBI coatings acid, alkanedioic acid and substituted isocyanatocycloalkane, compds with alkylamine (G) Polymer for P-16-0444A 4 06/26/2019 CBI (G) Amine salted polyurethane coatings (G) Carboxylic acids, unsaturated, hydrogenated polymers with (G) Polymer for substituted alkanediamine, alkanediol, substituted P-16-0445A 4 06/26/2019 CBI coatings alkylpropanoic acid, alkanedioic acid and substituted isocyanatocycloalkane, compds with alkylamine P-17-0007A 5 06/13/2019 CBI (S) Intermediate (G) Dialkyl 7,10-dioxa, dithiahexadeca diene (G) Substituted carboxylic acid, polymer with 2,4-diisocyanato- (G) Adhesive for 1-methylbenzene, hexanedioic acid, alpha-hydro-omega- P-17-0239A 6 06/11/2019 CBI open non-descriptive -

SAFETY DATA SHEET Sodium Persulfate
SAFETY DATA SHEET Sodium Persulfate SDS # : 7775-27-1 Revision date: 2021-02-17 Format: NA Version 1.06 1. PRODUCT AND COMPANY IDENTIFICATION Product Identifier Product Name Sodium Persulfate CAS-No 7775-27-1 Synonyms Sodium Peroxydisulfate; Disodium Peroxydisulfate; Peroxydisulfuric acid, disodium salt; Peroxydisulfuric acid, sodium salt. Recommended use of the chemical and restrictions on use Recommended Use: Polymerization initiator; Etchant and cleaner for printed circuit boards; Hair bleaching formulations; Secondary oil recovery; Oxidizing agent for a variety of organic reactions. Restrictions on Use No uses to be advised against were identified. Manufacturer/Supplier PeroxyChem LLC 2005 Market Street Suite 3200 Philadelphia, PA 19103 Phone: +1 267/ 422-2400 (General Information) E-Mail: [email protected] Emergency telephone numbers For leak, fire, spill or accident emergencies, call: 1 800 / 424 9300 (CHEMTREC - U.S.A.) 1 703 / 527 3887 (CHEMTREC - Collect - All Other Countries) +1 303/ 389-1409 (Medical - U.S. - Call Collect) Page 1 / 10 Sodium Persulfate SDS # : 7775-27-1 Revision date: 2021-02-17 Version 1.06 2. HAZARDS IDENTIFICATION Classification OSHA Regulatory Status This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200) Acute toxicity - Oral Category 4 Skin corrosion/irritation Category 2 Serious eye damage/eye irritation Category 2B Respiratory sensitization Category 1 Skin sensitization Category 1 Specific target organ toxicity (single exposure) Category 3 Oxidizing Solids -

19650003847.Pdf
NATIONAL BUREAU OF STANDARDS REPO RT 8595 PRELIMINARY REPORT ON A SURVEY OF THERMODYNAMIC PROPERTIES OF THE COMPOUNDS OF THE ELEMENTS CHNOPS i N65 2 CACC£S$IO NUMB£:R) CTHRU) ~ ___f£ / I- (PAGES) (CdOEI ::; ~ CIU - d -P?02c2/ :33 (NASA CR OR TMX OR AD NUMBER) (CATEQORY) Progress Report for the Period 1 August to 31 October 1964 to National Aeronautics and Space Administration GPO PRICE $ _____ OTS PRICE(S) $ 1 November 1964 Hard copy (He) of· cJV r Microfiche (MF) __....;... : .:::.j~I?J~_ <@> U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARD S The ational Bureau of Standards is a principal focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. It responsibilities include development and main tena nce of the national stand· ards of measurement, and the provisions of means for making measurements consi tent with those standard ; determination of physical constants and properties of materials; development of methods for testing materials, mechanisms, and structures, and making such tests as may be nece sary, particu larl: for ~overn ment agencies; cooperation in the establi hment of standard practices for incorpora tion in codes and specifications; advisory service to government agencies on scientific and technical problems; invention and development of device to serve special needs of the Government: assi tance to indus! r). business. and consumers in the development and acceptance of commercial tandard and simplified trade practice recommendations; administration of programs in cooperation with United tates bu iness groups and standards organizations for the development of international standard of practice; and maintenance of a clearinghouse for the collection and dis emination of scientific, tech nical. -

Organic Chemistry of Explosives
JWBK121-FM October 11, 2006 21:10 Char Count= 0 Organic Chemistry of Explosives Dr. Jai Prakash Agrawal CChem FRSC (UK) Former Director of Materials Defence R&D Organisation DRDO House, New Delhi, India email: [email protected] Dr. Robert Dale Hodgson Consultant Organic Chemist, Syntech Chemical Consultancy, Morecambe, Lancashire, UK Website: http://www.syntechconsultancy.co.uk email: [email protected] iii JWBK121-FM October 11, 2006 21:10 Char Count= 0 iii JWBK121-FM October 11, 2006 21:10 Char Count= 0 Organic Chemistry of Explosives i JWBK121-FM October 11, 2006 21:10 Char Count= 0 ii JWBK121-FM October 11, 2006 21:10 Char Count= 0 Organic Chemistry of Explosives Dr. Jai Prakash Agrawal CChem FRSC (UK) Former Director of Materials Defence R&D Organisation DRDO House, New Delhi, India email: [email protected] Dr. Robert Dale Hodgson Consultant Organic Chemist, Syntech Chemical Consultancy, Morecambe, Lancashire, UK Website: http://www.syntechconsultancy.co.uk email: [email protected] iii JWBK121-FM October 11, 2006 21:10 Char Count= 0 Copyright C 2007 John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England Telephone (+44) 1243 779777 Email (for orders and customer service enquiries): [email protected] Visit our Home Page on www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1T 4LP, UK, without the permission in writing of the Publisher. -

Bnl Chemical Storage Table
The only official copy of this document is on-line at the SBMS website. Before using a printed copy, verify that it is current by checking the effective date. BNL CHEMICAL STORAGE RECOMMENDATIONS This information is adapted from published references for the most common chemicals at BNL. It presents well known compatibility and storage issues but is not an inclusive list of chemicals, hazards, or compatibility issues. For guidance on chemicals not listed on this table, consult the Material Safety Data Sheet, the BNL Chemical Hygiene Officer or your Environmental Safety & Health Representative. The information provided in these tables is to serve as guidance for common work settings and may be modified as appropriate based on the quantity, container type, storage containment & safety apparatus, and work conditions. BNL CHEMICAL STORAGE INFORMATION FOR TABLE 1: COMPRESSED GASES IN CYLINDERS RECOMMENDED COLOR-CODING SYSTEM TABLE 2A: FOR SOLIDS AND LIQUIDS CONTAINER STORAGE BNL CHEMICAL STORAGE INFORMATION FOR TABLE 2B: CONTAINERS OF SOLIDS AND LIQUIDS 12.0/12029e011.pdf 1 of 22 (02/2011) The only official copy of this document is on-line at the SBMS website. Before using a printed copy, verify that it is current by checking the effective date. TABLE 1: BNL CHEMICAL STORAGE INFORMATION FOR COMPRESSED GASES IN CYLINDERS Note: See the Compressed Gas Cylinders and Related Systems Subject Area for additional information and requirements. Global Type Examples Harmonization Storage Symbol Can be stored with all CORROSIVE other classes unless the -

New York City Department of Environmental Protection Community Right-To-Know: List of Hazardous Substances
New York City Department of Environmental Protection Community Right-to-Know: List of Hazardous Substances Updated: 12/2015 Definitions SARA = The federal Superfund Amendments and Reauthorization Act (enacted in 1986). Title III of SARA, known as the Emergency Planning and Community Right-to-Act, sets requirements for hazardous chemicals, improves the public’s access to information on chemical hazards in their community, and establishes reporting responsibilities for facilities that store, use, and/or release hazardous chemicals. RQ = Reportable Quantity. An amount entered in this column indicates the substance may be reportable under §304 of SARA Title III. Amount is in pounds, a "K" represents 1,000 pounds. An asterisk following the Reporting Quantity (i.e. 5000*) will indicate that reporting of releases is not required if the diameter of the pieces of the solid metal released is equal to or exceeds 100 micrometers (0.004 inches). TPQ = Threshold Planning Quantity. An amount entered in this column reads in pounds and indicates the substance is an Extremely Hazardous Substance (EHS), and may require reporting under sections 302, 304 & 312 of SARA Title III. A TPQ with a slash (/) indicates a "split" TPQ. The number to the left of the slash is the substance's TPQ only if the substance is present in the form of a fine powder (particle size less than 100 microns), molten or in solution, or reacts with water (NFPA rating = 2, 3 or 4). The TPQ is 10,000 lb if the substance is present in other forms. A star (*) in the 313 column= The substance is reportable under §313 of SARA Title III. -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G. -

The Following List Contains the Material Safety Data Sheets You Requested
The following list contains the Material Safety Data Sheets you requested. Please scoll down to view the requested MSDS(s). Product MSDS Distributor Format Language Quantity 2429700 2084769 Hach Company ROWGHS English 1 2429700 2106069 Hach Company ROWGHS English 1 __________________________________________________________________________________________________________ Total Enclosures: 2 Obtained by Global Safety Management, www.globalsafetynet.com, (877) 683-7460 World Headquarters Hach Company P.O.Box 389 MSDS No: M00039 Loveland, CO USA 80539 (970) 669-3050 SAFETY DATA SHEET _____________________________________________________________________________ 1. CHEMICAL PRODUCT AND COMPANY IDENTIFICATION Product Name: Potassium Persulfate Catalog Number: 2084769 Hach Company Emergency Telephone Numbers: P.O.Box 389 (Medical and Transportation) Loveland, CO USA 80539 (303) 623-5716 24 Hour Service (970) 669-3050 (515)232-2533 8am - 4pm CST MSDS Number: M00039 Chemical Name: Peroxydisulfuric Acid, Dipotassium Salt CAS Number: 7727-21-1 Additional CAS No. (for hydrated forms): Not applicable Chemical Formula: K2S2O8 Chemical Family: Oxidizing Agents Intended Use: Laboratory Use _____________________________________________________________________________ 2. HAZARDS IDENTIFICATION GHS Classification: Hazard categories: Oxidizing Solids: Ox. Sol. 3 Acute Toxicity: Acute Tox. 4-Orl Skin Corrosion/Irritation: Skin Irrit. 2 Respiratory or Skin Sensitization: Skin Sens.1 Serious Eye Damage/Eye Irritation:Eye Irrit. 2 Respiratory or Skin Sensitization: Resp. Sens.1 Specific Target Organ Toxicity - Single Exposure: STOT SE 3 GHS Label Elements: DANGER Hazard statements: May intensify fire; oxidiser. Harmful if swallowed. Causes skin irritation. May cause an allergic skin reaction. Causes serious eye irritation. May cause allergy or asthma symptoms or breathing difficulties if inhaled. May cause respiratory irritation. Precautionary statements: Wear protective gloves / protective clothing / eye protection / face protection. -
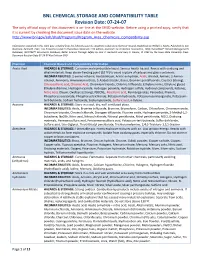
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers