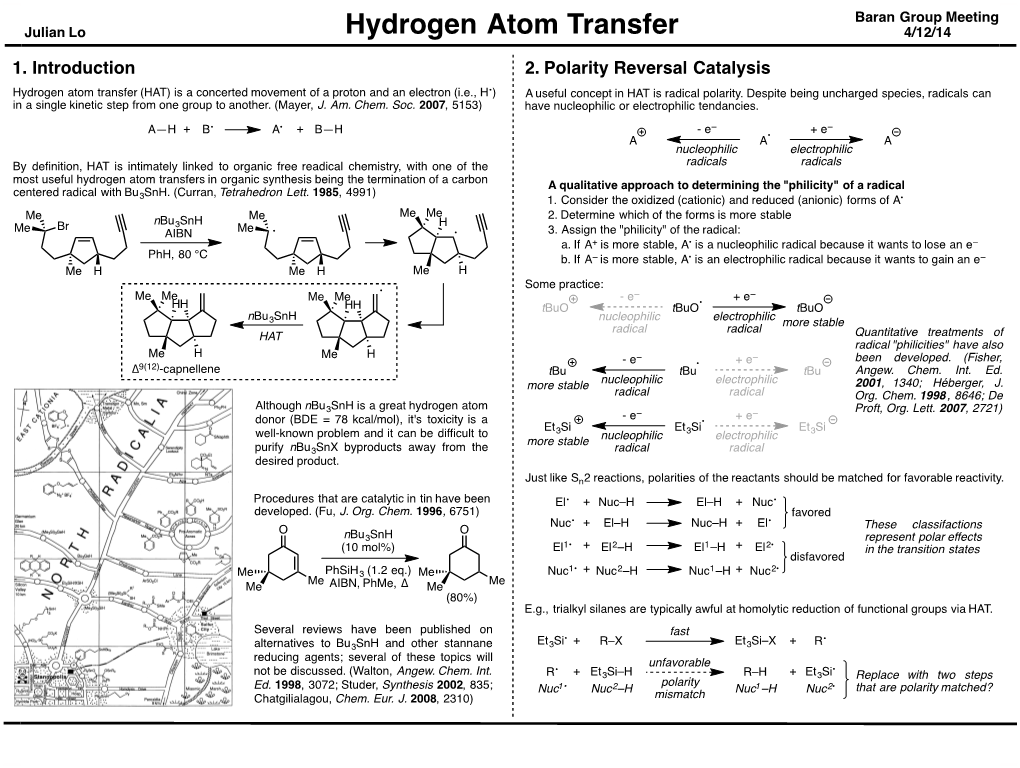
Hydrogen Atom Transfer 4/12/14
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Solid-State Structures of the Covalent Hydrides Germane and Stannane
Edinburgh Research Explorer Solid-state structures of the covalent hydrides germane and stannane Citation for published version: Maley, IJ, Brown, DH, Ibberson, RM & Pulham, CR 2008, 'Solid-state structures of the covalent hydrides germane and stannane', Acta Crystallographica Section B - Structural Science, vol. 64, no. Pt 3, pp. 312-7. https://doi.org/10.1107/S0108768108010379 Digital Object Identifier (DOI): 10.1107/S0108768108010379 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Acta Crystallographica Section B - Structural Science Publisher Rights Statement: Copyright © 2008 International Union of Crystallography; all rights reserved. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 electronic reprint Acta Crystallographica Section B Structural Science, Crystal Engineering and Materials ISSN 2052-5192 Solid-state structures of the covalent hydrides germane and stannane Iain J. Maley, Daniel H. Brown, Richard M. Ibberson and Colin R. Pulham Acta Cryst. (2008). B64, 312–317 Copyright c International Union of Crystallography Author(s) of this paper may load this reprint on their own web site or institutional repository provided that this cover page is retained. -

Stannane 2629
TRIMETHPLSTANNANE 2629 [CONTRIBUTION FROM THE CHEWCAL LABORATORY OF CLARK UNIVERSITY, I, 161 THE PREPARATION AND PROPERTIES OF TRIMETHYL- STANNANE BY CHARLESA. KRAUSAUD WILLARDTu'. GREER Recci\ed .4ugiist 11 1922 Introduction.-The hydrogen derivatives of the various elements decreasc in stability as we proceed from the elements of the halogen group to the elements of higher negative 1-alence,and as wc proceed from elements of lower to elements of higher atomic weight. A11 the elements of the halogen group give stable hydrides at ordinary temperatures. The hydrides of the higher member4 of the oxygen group have comparatively little stability, while in the case of the nitrogen group only nitrogen and phosphorus form comparatively stable normal hydrides. Of the fourth group of elements, carbon and silicon form stable hydrides, but the higher members form hydrides possessing little stability. It is only recently that the existence of hydrides o€ tin, lend and bismuth has been estab- lished.' ff one or more of the 111 drogens of a hydride is replaced by an alkyl or am1 group, the stability of the resulting compound is greatly increased over that of the hydride. Kearly all the common elements yield stable compounds of this type when all the hydrogens are substituted in this way. Tlie metallo-organic compounds of the elements of the fourth group exhibit unusual stability, corresponding somewhat to the exceptional stability of methane, the first member of the group. The tetra-alkyl tin compounds are, on the whole, very stable substances. It might be expected, therefore, that, in the case of tin if only a portion of the hydrogens mere wbstituted by alkyl groups, the resulting compound would exhibit a con- siclt~abledegree of stability. -

High Pressure Stabilization and Emergent Forms of Pbh4
week ending PRL 107, 037002 (2011) PHYSICAL REVIEW LETTERS 15 JULY 2011 High Pressure Stabilization and Emergent Forms of PbH4 Patryk Zaleski-Ejgierd,1 Roald Hoffmann,2 and N. W. Ashcroft1 1Laboratory of Atomic and Solid State Physics and Cornell Center for Materials Research, Clark Hall, Cornell University, Ithaca, New York 14853-2501, USA 2Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, New York 14853-1301, USA (Received 17 October 2010; revised manuscript received 9 March 2011; published 11 July 2011; corrected 20 July 2011) A wide decomposition pressure range of 132 GPa is predicted for PbH4 above which it emerges in very different forms compared with its group-14 congeners. This triply Born-Oppenheimer system is a nonmolecular, three-dimensional, metallic alloy, despite a prominent layered structure. A significant number of enthalpically near-degenerate structures, with exceedingly small energy barriers for distortions, and characteristic instabilities in the phonon spectra suggest that even at very high pressures PbH4 may exhibit both metallic and liquidlike properties and sublattice or even full melting. DOI: 10.1103/PhysRevLett.107.037002 PACS numbers: 74.70.Ad, 74.62.Fj Among group-14 hydrides, the lead-hydrogen system is As starting points and guides in a structure search, we special, for it clearly combines one of the heaviest (Pb) used a number of geometries previously reported for group- elements with the lightest (H). The attendant mass ratio 14 tetrahydrides, EH4, as well as other structures in which (207:1) immediately implies a distinct separation of time lead is two- to 16-fold coordinated by hydrogen, and with scales for the ensuing dynamics, in very much the way that up to Z ¼ 4 formula units per cell. -

Select Reactions of Organoboranes and Organostannanes
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2012 Select Reactions of Organoboranes and Organostannanes David W. Blevins [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Organic Chemistry Commons Recommended Citation Blevins, David W., "Select Reactions of Organoboranes and Organostannanes. " PhD diss., University of Tennessee, 2012. https://trace.tennessee.edu/utk_graddiss/1412 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by David W. Blevins entitled "Select Reactions of Organoboranes and Organostannanes." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. George W. Kabalka, Major Professor We have read this dissertation and recommend its acceptance: Shane Foister, Craig Barnes, Kimberly Gwinn Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Select Reactions of Organoboranes and Organostannanes A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville David W. Blevins August 2012 DEDICATION I dedicate this work to Lisa, Preston, and Clarissa Blevins. ii ACKNOWLEDGEMENTS I would like to thank Dr. -

Hydrides at High Pressures; an Evaluation of "Chemical Precompression" on the Way to Metallic Hydrogen
Hydrides at High Pressures; An Evaluation of "Chemical Precompression" on the way to Metallic Hydrogen by Paulina Gonzalez Morelos This thesis/dissertation document has been electronically approved by the following individuals: Hoffmann,Roald (Chairperson) Lee,Stephen (Minor Member) Chan,Garnet (Minor Member) HYDRIDES AT HIGH PRESSURES; AN EVALUATION OF “CHEMICAL PRECOMPRESSION” ON THE WAY TO METALLIC HYDROGEN A Dissertation Presented to the Faculty of the Graduate School of Cornell University in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Paulina Gonzalez Morelos August 2010 c 2010 Paulina Gonzalez Morelos ALL RIGHTS RESERVED HYDRIDES AT HIGH PRESSURES; AN EVALUATION OF “CHEMICAL PRECOMPRESSION” ON THE WAY TO METALLIC HYDROGEN Paulina Gonzalez Morelos, Ph.D. Cornell University 2010 In this dissertation, we present a theoretical study of compounds rich in hydro- gen, focusing on their crystal structure and potential metallization as pressure is applied. Chapter 1 is an introduction to metallic hydrogen; it reviews previous stud- ies and suggests a new way of analyzing the geometric changes that ensue as pressure is applied. Chapter 2 describes the search for structures for SnH4, us- ing chemical intuition and random search algorithms. Chapter 3 deals with the general problem of segregation which arises as SnH4, a thermodynamically metastable (positive heat of formation from Sn and 2H2) but kinetically persis- tent compound, is compressed. Chapter 3 is a complement to Chapter 2, as layered structures arise naturally in the low pressure regime, prompting us to explore the subject. Chapter 4 begins an ambitious project which strives to understand the elec- tronic structure and properties of metallic hydrides of the form MHx, where M= Si, Sn and W, and x = 4, 6, 8, 10, 12. -

This Table Gives the Standard State Chemical Thermodynamic Properties of About 2500 Individual Substances in the Crystalline, Liquid, and Gaseous States
STANDARD THERMODYNAMIC PROPERTIES OF CHEMICAL SUBSTANCES This table gives the standard state chemical thermodynamic properties of about 2500 individual substances in the crystalline, liquid, and gaseous states. Substances are listed by molecular formula in a modified Hill order; all substances not containing carbon appear first, followed by those that contain carbon. The properties tabulated are: DfH° Standard molar enthalpy (heat) of formation at 298.15 K in kJ/mol DfG° Standard molar Gibbs energy of formation at 298.15 K in kJ/mol S° Standard molar entropy at 298.15 K in J/mol K Cp Molar heat capacity at constant pressure at 298.15 K in J/mol K The standard state pressure is 100 kPa (1 bar). The standard states are defined for different phases by: • The standard state of a pure gaseous substance is that of the substance as a (hypothetical) ideal gas at the standard state pressure. • The standard state of a pure liquid substance is that of the liquid under the standard state pressure. • The standard state of a pure crystalline substance is that of the crystalline substance under the standard state pressure. An entry of 0.0 for DfH° for an element indicates the reference state of that element. See References 1 and 2 for further information on reference states. A blank means no value is available. The data are derived from the sources listed in the references, from other papers appearing in the Journal of Physical and Chemical Reference Data, and from the primary research literature. We are indebted to M. V. Korobov for providing data on fullerene compounds. -

1 Draft Chemicals (Management and Safety)
Draft Chemicals (Management and Safety) Rules, 20xx In exercise of the powers conferred by Sections 3, 6 and 25 of the Environment (Protection) Act, 1986 (29 of 1986), and in supersession of the Manufacture, Storage and Import of Hazardous Chemical Rules, 1989 and the Chemical Accidents (Emergency Planning. Preparedness and Response) Rules, 1996, except things done or omitted to be done before such supersession, the Central Government hereby makes the following Rules relating to the management and safety of chemicals, namely: 1. Short Title and Commencement (1) These Rules may be called the Chemicals (Management and Safety) Rules, 20xx. (2) These Rules shall come into force on the date of their publication in the Official Gazette. Chapter I Definitions, Objectives and Scope 2. Definitions (1) In these Rules, unless the context otherwise requires (a) “Act” means the Environment (Protection) Act, 1986 (29 of 1986) as amended from time to time; (b) “Article” means any object whose function is determined by its shape, surface or design to a greater degree than its chemical composition; (c) “Authorised Representative” means a natural or juristic person in India who is authorised by a foreign Manufacturer under Rule 6(2); (d) “Chemical Accident” means an accident involving a sudden or unintended occurrence while handling any Hazardous Chemical, resulting in exposure (continuous, intermittent or repeated) to the Hazardous Chemical causing death or injury to any person or damage to any property, but does not include an accident by reason only -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G. -

Studies of the Hydrides of the Heavier Group 14 Elements
Studies of the Hydrides of the Heavier Group 14 Elements Daniel Hugh Brown A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy to the University of Edinburgh 1998 Declaration This thesis has been composed by myself and it has not been submitted in any previous application for a degree. The work reported within was executed by myself, unless otherwise stated. May 1998 1 Abstract The chemistry of some simple tin hydrides (CH3)4_ SnH (x = 1-4) has been investigated with particular reference to their reactivity towards the reagents NaH, KH, alkyl lithium compounds, and the ylids Me 3P C H2 and Ph3PCH2 . It has been found that the reaction between the stannanes and alkali metal hydrides or the ylids produces salts containing the respective stannyl anions, [Sn(CHa)sH] (x = 0-3). These salts have been characterised by low tem- perature infrared spectroscopy, the results of which are in good agreement with the results of ab initio calculations. In contrast, reactions involving alkyl lithium compounds proceed by transmetallation with elimination of LiH and alkylation of the tin centre. The technique of in situ crystal growth at low temperatures has been used to obtain X-ray crystal structures of Me 3P C H2 , Me2Sn H2 and Me3Sn H. The structure of the ylid in the solid phase has been shown to resemble the calculated structure of a transition state involving rotation of the CH 2 group about the P-C aids. The reaction between monochlorostannane and sodium metal produced small quantities of Sn 2H6, detected by mass spectrometry. -

IONIZATION ENERGIES of GAS-PHASE MOLECULES Sharon G
IONIZATION ENERGIES OF GAS-PHASE MOLECULES Sharon G. Lias This table presents values for the first ionization energies (IP) of approximately 1000 molecules and atoms. Substances are listed by molecular formula in the modified Hill order (see introduction). Values enclosed in parentheses are considered not to be well established. Data appearing in the 1988 reference, were updated in 1996 for inclusion in the database of ionization energies available at the Internet site of the Standard Reference Data program of the National Institute of Standards and Technology (http://webbook.nist.gov). The list appearing here includes these updates. ∆ The list also includes values for enthalpies of formation of the ions at 298 K, fHion, given according to the ion convention used by mass ∆ spectrometrists; to convert these values to the electron convention used by thermodynamicists, add 6 kJ/mol. Details on the calculation of fHion as well as data for a much larger number of molecules, may be found in the reference and on the Internet site. REFERENCE Lias, S.G., Bartmess, J.E., Liebman, J.F., Holmes, J.L., Levin, R.D., and Mallard, W.G., Gas-Phase Ion and Neutral Thermochemistry, J. Phys. Chem. Ref. Data, Vol. 17, Suppl. No. 1, 1988. ∆ Mol. f Hion Form. Name IP/eV kJ/mol Substances not containing carbon Ac Actinium 5.17 905 Ag Silver 7.57624 1016 AgCl Silver(I) chloride (≤ 10.08) ≤ 1065 AgF Silver(I) fluoride (11.0 ± 0.3) 1071 Al Aluminum 5.98577 905 AlBr Aluminum monobromide (9.3) 913 AlBr3 Aluminum tribromide (10.4) 593 AlCl Aluminum monochloride 9.4 -

Berkeley, California ...J V1 \Jj -C
Lawrence Berkeley National Laboratory Recent Work Title REACTIONS OF SILANE, GERMANE AND STANNANE WITH METAL-AND AMIDE-AMMONIA SOLUTIONS Permalink https://escholarship.org/uc/item/1f52s7rf Authors Rustad, Douglas S. Jolly, William L. Publication Date 1967-04-01 eScholarship.org Powered by the California Digital Library University of California UCRL-17534 t..~ ~. University of California Ernest O. lawrence Radiation Laboratory REACTIONS OF SILANE, GERMANE AND STANNANE WITH METAL- AND AMIDE-AMMONIA SOLUTIONS Douglas S. Rustad and William L. Jolly April 1967 C n 1'J \, Berkeley, California ...J V1 \jJ -C 1\ -t Y DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. , Submitted to Inorganic Chemistry UCRL-17534 Preprint UNIVERSITY OF CALIFORNIA .