High Pressure Stabilization and Emergent Forms of Pbh4
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
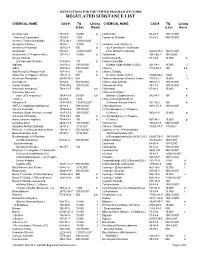
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Germane Facts About Germanium Sesquioxide: I. Chemistry and Anticancer Properties
THE JOURNAL OF ALTERNATIVE AND COMPLEMENTARY MEDICINE Volume 10, Number 2, 2004, pp. 337–344 ©Mary Ann Liebert, Inc. Germane Facts About Germanium Sesquioxide: I. Chemistry and Anticancer Properties BONNIEJ. KAPLAN, Ph.D., 1 W. WESLEYPARISH, Ph.D., 2 G. MERRILLANDRUS, Ph.D., 2 J. STEVENA. SIMPSON, Ph.D., M.D., 3 and CATHERINEJ. FIELD, Ph.D., R.D. 4 ABSTRACT This paper reviews the history, chemistry, safety, toxicity, and anticancer effects of the organogermanium compound bis (2-carboxyethylgermanium) sesquioxide (CEGS). A companion review follows, discussing the inaccuracies in the scientific record that have prematurely terminated research on clinical uses of CEGS. CEGS is a unique organogermanium compound first made by Mironov and coworkers in Russia and, shortly there- after, popularized by Asai and his colleagues in Japan. Low concentrations of germanium occur in nearly all soils, plants and animal life; natural occurrence of the CEGS form is postulated but not yet demonstrated. The literature demonstrating its anticancer effect is particularly strong: CEGS induces interferon- g (IFN-g), en- hances natural killer cell activity, and inhibits tumor and metastatic growth—effects often detectable after a single oral dose. In addition, oral consumption of CEGS is readily assimilated and rapidly cleared from the body without evidence of toxicity. Given these findings, the absence of human clinical trials of CEGS is un- expected. Possible explanations of why the convincing findings from animal research have not been used to support clinical trials are discussed. Clinical trials on CEGS are recommended. INTRODUCTION bispropionic acid; 3-oxygermylpropionic acid polymer; poly- trans-(2-carboxyethyl) germasesquioxane); proxigerma- n general, dietary supplements are an underinvestigated nium; repagermanium; and Serocion. -

Germane 99.99+%
GEG5001 - GERMANE 99.99+% GERMANE 99.99+% Safety Data Sheet GEG5001 Date of issue: 01/05/2015 Version: 1.0 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Product form : Substance Physical state : Gas Substance name : GERMANE 99.99+% Product code : GEG5001 Formula : GeH4 Synonyms : MONOGERMANE; GERMANIUM HYDRIDE; GERMANIUM TETRAHYDRIDE Chemical family : GERMANE 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Chemical intermediate For research and industrial use only 1.3. Details of the supplier of the safety data sheet GELEST, INC. 11 East Steel Road Morrisville, PA 19067 USA T 215-547-1015 - F 215-547-2484 - (M-F): 8:00 AM - 5:30 PM EST [email protected] - www.gelest.com 1.4. Emergency telephone number Emergency number : CHEMTREC: 1-800-424-9300 (USA); +1 703-527-3887 (International) SECTION 2: Hazards identification 2.1. Classification of the substance or mixture Classification (GHS-US) Flam. Gas 1 H220 Liquefied gas H280 Acute Tox. 2 (Inhalation:gas) H330 Eye Irrit. 2A H319 STOT SE 3 H335 Full text of H-phrases: see section 16 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS02 GHS04 GHS06 GHS07 Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H220 - Extremely flammable gas H280 - Contains gas under pressure; may explode if heated H319 - Causes serious eye irritation H330 - Fatal if inhaled H335 - May cause respiratory irritation Precautionary statements (GHS-US) : P284 - [In case of inadequate ventilation] wear respiratory protection P280 - Wear protective gloves/protective clothing/eye protection/face protection P260 - Do not breathe gas P264 - Wash hands thoroughly after handling P310 - Immediately call a doctor P210 - Keep away from heat, open flames, sparks. -

Solid-State Structures of the Covalent Hydrides Germane and Stannane
Edinburgh Research Explorer Solid-state structures of the covalent hydrides germane and stannane Citation for published version: Maley, IJ, Brown, DH, Ibberson, RM & Pulham, CR 2008, 'Solid-state structures of the covalent hydrides germane and stannane', Acta Crystallographica Section B - Structural Science, vol. 64, no. Pt 3, pp. 312-7. https://doi.org/10.1107/S0108768108010379 Digital Object Identifier (DOI): 10.1107/S0108768108010379 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Acta Crystallographica Section B - Structural Science Publisher Rights Statement: Copyright © 2008 International Union of Crystallography; all rights reserved. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 electronic reprint Acta Crystallographica Section B Structural Science, Crystal Engineering and Materials ISSN 2052-5192 Solid-state structures of the covalent hydrides germane and stannane Iain J. Maley, Daniel H. Brown, Richard M. Ibberson and Colin R. Pulham Acta Cryst. (2008). B64, 312–317 Copyright c International Union of Crystallography Author(s) of this paper may load this reprint on their own web site or institutional repository provided that this cover page is retained. -

Germane Interim AEGL Document
1 2 3 4 ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 5 FOR 6 GERMANE 7 (CAS Reg. No. 7782-65-2) 8 9 GeH4 10 11 12 13 14 15 16 17 INTERIM 18 19 GERMANE Interim: 09-2009/ Page 2 of 31 1 2 ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 3 FOR 4 GERMANE 5 (CAS Reg. No. 7782-65-2) 6 7 8 9 10 INTERIM 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 GERMANE Interim: 09-2009/ Page 3 of 31 1 PREFACE 2 3 Under the authority of the Federal Advisory Committee Act (FACA) P. L. 92-463 of 4 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous 5 Substances (NAC/AEGL Committee) has been established to identify, review and interpret 6 relevant toxicologic and other scientific data and develop AEGLs for high priority, acutely toxic 7 chemicals. 8 9 AEGLs represent threshold exposure limits for the general public and are applicable to 10 emergency exposure periods ranging from 10 minutes to 8 hours. Three levels C AEGL-1, 11 AEGL-2 and AEGL-3 C are developed for each of five exposure periods (10 and 30 minutes, 1 12 hour, 4 hours, and 8 hours) and are distinguished by varying degrees of severity of toxic effects. 13 The three AEGLs are defined as follows: 14 15 AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per 16 cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general 17 population, including susceptible individuals, could experience notable discomfort, irritation, or 18 certain asymptomatic, non-sensory effects. -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Digermane SDS 1
SAFETY DATA SHEET Prepared to U.S. OSHA, CMA, ANSI, Canadian WHMIS Standards, European Union CLP EC 1272/2008, REACH, and the Global Harmonization Standard 1. SECTION 1 – IDENTIFICATION OF THE SUBSTANCE/MIXTURE AND OF THE COMPANY/UNDERTAKING CHEMICAL NAME; CLASS: DIGERMANE SYNONYMS: Digerman; λ2-germanylidenegermanium; Germanium Hexahydride; Germanium Hydride, Germanium (III) Hydride, Germanium Trihydride CHEMICAL FAMILY: Hydride FORMULA: Ge 2H6 Docu ment Numbe r: 80 019 PR ODUCT USE: Various MANU FA CTUR ED/SU PPL IED FOR: SUPPLIER/MANUFACTURER'S NAME: AIR LIQUIDE AMERICA ADDRESS : 2700 Post Oak Drive Hous ton , TX 7705 6-822 9 EM AIL ADD RES S FOR PRODUC T INF ORM ATION: webmas ter.us @airliqu ide.co m EMERGENCY PHONE: CHEMTREC: (U.S., Canada) 1-800-424-9300 (24 hrs) (International) +703-527-3887 (collec t-24 hrs) BUSINESS PHONE: General MSDS Information: 1-713/896-2896 (8 am to 5 pm U.S. Central Time) Fax on Demand: 1-800/231-1366 ALL WHMIS required information is included in appropriate sections based on the ANSI Z400.1-2004 format. This product has been classified in accordance with the hazard criteria of the CPR and the MSDS contains all the information required by the CPR. The product is also classified per all applicable European Union CLP EC 1272/2008 and the Global Harmonization Standard. TSCA Status: This material is not included in the TSCA Inventory. In accordance with the conditions listed in 40 CFR 720.36 and 721.47, this product must be used only for research and development, pharmaceutical manufacture, or export. -

United States Patent to 11, 4,018,606 Contois Et Al
United States Patent to 11, 4,018,606 Contois et al. 45 Apr. 19, 1977 (54) ORGANIC AZO PIGMENT SENSITIZERS FOR PHOTOCONDUCTIVE LAYERS R ph C 2 x (75) Inventors: Lawrence E. Contois; Joseph Y. R2 NEN-C Kaukeinen; Stephen Michel; Thomas C M. Plutchak, all of Rochester, N.Y. R R (73) Assignee: Eastman Kodak Company, wherein X consists of the atoms necessary to complete Rochester, N.Y. a naphthalene, anthracene, or OH (22 Filed: May 3, 1974 C 2 N. -C N N (21) Appl. No.: 466,658 =N R (52) U.S. Cl. ...................................... 96/1.7; 96/1.6 ring; (51) Int. Cl”.......................................... G03G 5/09 (58) Field of Search ........................... 96/1, 1.5, 1.6 R', R', and R are hydrogen, halogen, alkoxy, NO, alkyl, SOH or alkali metal salts thereof, (56) References Cited O O UNITED STATES PATENTS rCNH NO, and -CNH 3,384,632 5/1968 Solodar ................................ 96/1.6 3,622,341 8/1969 Lee ....................................... 96/1.6 and COOH or alkali metal salts thereof, and Rand R8 3,684,548 8/1972 Contois ............................... 96/1.6 can comprise the atoms necessary to complete a 3,775,105 l/1973 Kukla ................................... 96/1.6 phenyl, naphthyl or anthryl ring; and R is selected from FOREIGN PATENTS OR APPLICATIONS the group consisting of 1,370,197 10/1974 United Kingdom .................. 96/1.5 O O H il H Primary Examiner-David Klein 8-i-O-No, 8-N-O). or COOM Assistant Examiner-John L. Goodrow Attorney, Agent, or Firm-Arthur H. Rosenstein where M is alkyl, alkali or alkaline earth metal are useful as sensitizers for photoconductive compositions in electrophotographic processes. -

40 CFR Ch. I (7–1–14 Edition) Pt. 68, App. A
Pt. 68, App. A 40 CFR Ch. I (7–1–14 Edition) under subpart G of this part as required (j) Nothing in this section shall pre- by the final determination, and sub- clude, limit, or interfere in any way mits the revised RMP as required with the authority of EPA or the state under § 68.150. to exercise its enforcement, investiga- (i) The public shall have access to the tory, and information gathering au- preliminary determinations, responses, thorities concerning this part under and final determinations under this the Act. section in a manner consistent with § 68.210. APPENDIX A TO PART 68—TABLE OF TOXIC ENDPOINTS [As defined in § 68.22 of this part] Toxic end- CAS No. Chemical name point (mg/L) 107–02–8 ............. Acrolein [2-Propenal] ................................................................................................................ 0 .0011 107–13–1 ............. Acrylonitrile [2-Propenenitrile] ................................................................................................... 0.076 814–68–6 ............. Acrylyl chloride [2-Propenoyl chloride] ..................................................................................... 0 .00090 107–18–6 ............. Allyl alcohol [2-Propen-1-ol] ...................................................................................................... 0 .036 107–11–9 ............. Allylamine [2-Propen-1-amine] ................................................................................................. 0 .0032 7664–41–7 ........... Ammonia (anhydrous) ............................................................................................................. -

HIGH HAZARD GAS Review Date: 09/23/2019
University of Pittsburgh EH&S Guideline Number: 04-021 Safety Manual Subject: Effective Date: 04/19/2017 Page 1 of 9 HIGH HAZARD GAS Review Date: 09/23/2019 STORAGE AND USE OF HIGH HAZARD GAS 1. Definition of High Hazard (HH) Gases For these guidelines, any gas meeting one or more of the following definitions based on International Fire Code (IFC) and National Fire Protection Association (NFPA) standards: 1.1. Flammable gas – a material that is a gas at 68ºF (20ºC) or less at an absolute pressure of 14.7 psi (101.325 kPa) when in a mixture of 13% or less by volume with air, or that has a flammable range at an absolute pressure of 14.7 psi (101.325 kPa) with air of at least 12%, regardless of the lower limit 1.2. Pyrophoric gas – a gas with an autoignition temperature in air at or below 130ºF (54.4ºC) 1.3. Health Hazard 3 (HH3) gas – material that, under emergency conditions and according to the standards, can cause serious or permanent injury 1.4. Health Hazard 4 (HH4) gas – material that, under emergency conditions and according to the standards, can be lethal The storage and usage of a gas or gases meeting any of the above definitions must follow all applicable IFC and NFPA guidelines and the requirements outlined in this document. Consult EH&S for specific guidance on gas mixtures containing corrosive, flammable or poisonous gas components (ex. 1% carbon monoxide/nitrogen, 5% hydrogen sulfide/helium). 2. Notification Requirements Prior to Obtaining High Hazard Gases 2.1. -

Stannane 2629
TRIMETHPLSTANNANE 2629 [CONTRIBUTION FROM THE CHEWCAL LABORATORY OF CLARK UNIVERSITY, I, 161 THE PREPARATION AND PROPERTIES OF TRIMETHYL- STANNANE BY CHARLESA. KRAUSAUD WILLARDTu'. GREER Recci\ed .4ugiist 11 1922 Introduction.-The hydrogen derivatives of the various elements decreasc in stability as we proceed from the elements of the halogen group to the elements of higher negative 1-alence,and as wc proceed from elements of lower to elements of higher atomic weight. A11 the elements of the halogen group give stable hydrides at ordinary temperatures. The hydrides of the higher member4 of the oxygen group have comparatively little stability, while in the case of the nitrogen group only nitrogen and phosphorus form comparatively stable normal hydrides. Of the fourth group of elements, carbon and silicon form stable hydrides, but the higher members form hydrides possessing little stability. It is only recently that the existence of hydrides o€ tin, lend and bismuth has been estab- lished.' ff one or more of the 111 drogens of a hydride is replaced by an alkyl or am1 group, the stability of the resulting compound is greatly increased over that of the hydride. Kearly all the common elements yield stable compounds of this type when all the hydrogens are substituted in this way. Tlie metallo-organic compounds of the elements of the fourth group exhibit unusual stability, corresponding somewhat to the exceptional stability of methane, the first member of the group. The tetra-alkyl tin compounds are, on the whole, very stable substances. It might be expected, therefore, that, in the case of tin if only a portion of the hydrogens mere wbstituted by alkyl groups, the resulting compound would exhibit a con- siclt~abledegree of stability. -

Environmental Protection Agency § 68.130
Environmental Protection Agency § 68.130 include, but are not limited to, phys- TABLE 1 TO § 68.130—LIST OF REGULATED ical and chemical properties of the sub- TOXIC SUBSTANCES AND THRESHOLD QUAN- stance, such as vapor pressure; mod- TITIES FOR ACCIDENTAL RELEASE PREVEN- eling results, including data and as- TION—Continued sumptions used and model documenta- [Alphabetical Order—77 Substances] tion; and historical accident data, cit- ing data sources. Threshold Chemical name CAS No. quantity Basis for (h) Within 18 months of receipt of a (lbs) listing petition, the Administrator shall pub- Ammonia (anhy- 7664–41–7 10,000 a, b lish in the FEDERAL REGISTER a notice drous). either denying the petition or granting Ammonia (conc 7664–41–7 20,000 a, b the petition and proposing a listing. 20% or greater). Arsenous tri- 7784–34–1 15,000 b § 68.125 Exemptions. chloride. Arsine ................. 7784–42–1 1,000 b Agricultural nutrients. Ammonia used Boron trichloride 10294–34–5 5,000 b as an agricultural nutrient, when held [Borane, by farmers, is exempt from all provi- trichloro-]. Boron trifluoride 7637–07–2 5,000 b sions of this part. [Borane, trifluoro-]. § 68.126 Exclusion. Boron trifluoride 353–42–4 15,000 b compound with Flammable Substances Used as Fuel or methyl ether Held for Sale as Fuel at Retail Facilities. (1:1) [Boron, A flammable substance listed in Tables trifluoro [oxybis 3 and 4 of § 68.130 is nevertheless ex- [metane]]-, T-4-. Bromine .............. 7726–95–6 10,000 a, b cluded from all provisions of this part Carbon disulfide 75–15–0 20,000 b when the substance is used as a fuel or Chlorine .............