Select Reactions of Organoboranes and Organostannanes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mercury(II) Heterobimetallic Complexes Using a Hybrid Ligand, 1, 2-Bis(Diphenylphosphino) Ethane Monoxide
37 Sciencia Acta Xaveriana Volume 5 An International Science Journal No. 1 ISSN. 0976-1152 pp. 37-46 March 2014 Synthesis of organotin(IV) - mercury(II) heterobimetallic complexes using a hybrid ligand, 1, 2-bis(diphenylphosphino) ethane monoxide E. M. Mothia* and K. Panchantheswaranb a Department of Chemistry, Saveetha School of Engineering, Saveetha University, Thandalam, Chennai 602 105, India b School of Chemistry, Bharathidasan University, Tiruchirappalli 620 024, India *Email : [email protected] Abstract : The reaction of 1, 2-bis(diphenylphosphino)ethane monoxide, a hybrid ligand containing both soft (P) and hard (O) Lewis base centers in the same molecule, with organotin(IV) chloride and mercury(II) chloride has been carried out using dichloromethane as solvent. The products [HgCl2.(dppeO).R2SnCl2]n [R = Me (1), Ph (2) and CH2Ph (3)] have been obtained in excellent yields and are characterized using IR, 1H and 31P NMR spectroscopic methods. Based on the spectral data it is proposed that the structure may contain -Hg-P-O-Sn-O-P- Hg- polymeric array. Keywords : hybrid ligands, organotin, mercury, phosphine, phosphine oxide (Received : January 2014, Accepted February 2014) 38 Synthesis of organotin(IV) - mercury(II) heterobimetallic complexes using a hybrid ligand 1. Introduction Hybrid ligands are polydentate ligands that contain at least two different types of chemical functionality capable of binding to metal centers. These functionalities are often chosen to be very different from each other to increase the difference between their resulting interactions with metal centers and thereby contribute to chemoselectivity. Combining hard and soft donors in the same ligand has marked the evolution of different and contrasting chemistries, thus leading to novel and unprecedented properties for the resulting metal complexes.1An important class of such hybrid ligands are bis-phosphine monoxides (BPMOs) of the general formula R1R2P-Y-P(O)R3R4, where Y is a divalent spacer. -
Regulated Substance List
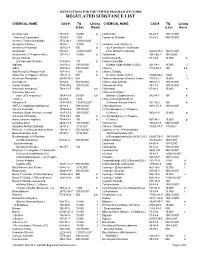
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Design and Synthesis of FRET-Based Boronic Acid Receptors to Detect Carbohydrate Clustering and Development of Diacylglycerol-Ba
University of Tennessee, Knoxville Trace: Tennessee Research and Creative Exchange Masters Theses Graduate School 12-2009 Design and Synthesis of FRET-Based Boronic Acid Receptors to Detect Carbohydrate Clustering and Development of Diacylglycerol-Based Lipid Probesto Investigate Lipid-Protein Binding Interactions Manpreet Kaur Cheema University of Tennessee - Knoxville Recommended Citation Cheema, Manpreet Kaur, "Design and Synthesis of FRET-Based Boronic Acid Receptors to Detect Carbohydrate Clustering and Development of Diacylglycerol-Based Lipid Probesto Investigate Lipid-Protein Binding Interactions. " Master's Thesis, University of Tennessee, 2009. https://trace.tennessee.edu/utk_gradthes/517 This Thesis is brought to you for free and open access by the Graduate School at Trace: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of Trace: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Manpreet Kaur Cheema entitled "Design and Synthesis of FRET-Based Boronic Acid Receptors to Detect Carbohydrate Clustering and Development of Diacylglycerol-Based Lipid Probesto Investigate Lipid-Protein Binding Interactions." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Master of Science, with a major in Chemistry. Michael Best, Major -

Chem 353: Grignard
GRIG.1 ORGANIC SYNTHESIS: BENZOIC ACID VIA A GRIGNARD REACTION TECHNIQUES REQUIRED : Reflux with addition apparatus, rotary evaporation OTHER DOCUMENTS Experimental procedure, product spectra INTRODUCTION In this experiment you will synthesise benzoic acid using bromobenzene to prepare a Grignard reagent, which is then reacted with carbon dioxide, worked-up and purified to give the acid. This sequence serves to illustrate some important concepts of practical synthetic organic chemistry : preparing and working with air and moisture sensitive reagents, the "work-up", extractions, apparatus set-up, etc. The synthesis utilises one of the most important type of reagents discussed in introductory organic chemistry, organometallic reagents. In this reaction, the Grignard reagent (an organomagnesium compound), phenylmagnesium bromide is prepared by reaction of bromobenzene with magnesium metal in diethyl ether (the solvent). The Grignard reagent will then be converted to benzoic acid via the reaction of the Grignard reagent with excess dry ice (solid CO2) followed by a "work-up" using dilute aqueous acid : The aryl (or alkyl) group of the Grignard reagent behaves as if it has the characteristics of a carbanion so it is a source of nucleophilic carbon. It is reasonable to represent the structure of the - + Grignard reagent as a partly ionic compound, R ....MgX. This partially-bonded carbanion is a very strong base and will react with acids (HA) to give an alkane: RH + MgAX RMgX + HA Any compound with suitably acidic hydrogens will readily donate a proton to destroy the reagent. Water, alcohols, terminal acetylenes, phenols and carboxylic acids are just some of the functional groups that are sufficiently acidic to bring about this reaction which is usually an unwanted side reaction that destroys the Grignard reagent. -

CO2 Derivatives of Molecular Tin Compounds. Part 1: † Hemicarbonato and Carbonato Complexes
inorganics Review CO2 Derivatives of Molecular Tin Compounds. Part 1: y Hemicarbonato and Carbonato Complexes Laurent Plasseraud Institut de Chimie Moléculaire de l’Université de Bourgogne (ICMUB), UMR-CNRS 6302, Université de Bourgogne Franche-Comté, 9 avenue A. Savary, F-21078 Dijon, France; [email protected] Paper dedicated to Professor Georg Süss-Fink on the occasion of his 70th birthday. y Received: 31 March 2020; Accepted: 24 April 2020; Published: 29 April 2020 Abstract: This review focuses on organotin compounds bearing hemicarbonate and carbonate ligands, and whose molecular structures have been previously resolved by single-crystal X-ray diffraction analysis. Most of them were isolated within the framework of studies devoted to the reactivity of tin precursors with carbon dioxide at atmospheric or elevated pressure. Alternatively, and essentially for the preparation of some carbonato derivatives, inorganic carbonate salts such as K2CO3, Cs2CO3, Na2CO3 and NaHCO3 were also used as coreagents. In terms of the number of X-ray structures, carbonate compounds are the most widely represented (to date, there are 23 depositions in the Cambridge Structural Database), while hemicarbonate derivatives are rarer; only three have so far been characterized in the solid-state, and exclusively for diorganotin complexes. For each compound, the synthesis conditions are first specified. Structural aspects involving, in particular, the modes of coordination of the hemicarbonato and carbonato moieties and the coordination geometry around tin are then described and illustrated (for most cases) by showing molecular representations. Moreover, when they were available in the original reports, some characteristic spectroscopic data are also given for comparison (in table form). -

Synthesis and Kinetics of Novel Ionic Liquid Soluble Hydrogen Atom Transfer Reagents
Synthesis and kinetics of novel ionic liquid soluble hydrogen atom transfer reagents Thomas William Garrard Submitted in total fulfilment of the requirements of the degree Doctor of Philosophy June 2018 School of Chemistry The University of Melbourne Produced on archival quality paper ORCID: 0000-0002-2987-0937 Abstract The use of radical methodologies has been greatly developed in the last 50 years, and in an effort to continue this progress, the reactivity of radical reactions in greener alternative solvents is desired. The work herein describes the synthesis of novel hydrogen atom transfer reagents for use in radical chemistry, along with a comparison of rate constants and Arrhenius parameters. Two tertiary thiol-based hydrogen atom transfer reagents, 3-(6-mercapto-6-methylheptyl)-1,2- dimethyl-3H-imidazolium tetrafluoroborate and 2-methyl-7-(2-methylimidazol-1-yl)heptane-2-thiol, have been synthesised. These are modelled on traditional thiol reagents, with a six-carbon chain with an imidazole ring on one end and tertiary thiol on the other. 3-(6-mercapto-6-methylheptyl)- 1,2-dimethyl-3H-imidazolium tetrafluoroborate comprises of a charged imidazolium ring, while 2- methyl-7-(2-methylimidazol-1-yl)heptane-2-thiol has an uncharged imidazole ring in order to probe the impact of salt formation on radical kinetics. The key step in the synthesis was addition of thioacetic acid across an alkene to generate a tertiary thioester, before deprotection with either LiAlH4 or aqueous NH3. Arrhenius plots were generated to give information on rate constants for H-atom transfer to a primary alkyl radical, the 5-hexenyl radical, in ethylmethylimidazolium bis(trifluoromethane)sulfonimide. -

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Organometallic Compounds
Chapter 11 Organometallic compounds Organometallics Reactions using organometallics Organometallic compounds Ch 11 #2 = comp’s containing a carbon-metal bond δ− δ+ C in organometallic comp’ds are nucleophilic. C in organic comp’d (like ROH, RNH , and RX) 2 δ+ δ are electrophilic. − due to ∆EN Ch 11 #3 in substitution reactions a carbon Nu: R-Li and R-MgX Ch 11 #4 (used to be) the two most common organometallics organolithium comp’ds BuLi = an alkyl lithium organomagnesium comp’ds = Grignard reagents 1912 Nobel Prize R, Ar, vinyl all possible; Br (as X) popular Ether (solvent) coordinates Mg, stabilizing it. Reactions of R-Li and R-MgX Ch 11 #5 reacts like a carbanion C:– ~ a C Nu: reactions as C Nu: (like SN(2)) nucleophilic addition to carbonyls ~ more often Chapt 16 Ch 11 #6 R-Li and R-MgX are very strong B: how strong? pKa? − − react even with very weak acid stronger than OH? NH2? useful for preparing deuterated HC Storage and reaction must be acid- and moisture-free. Transmetal(l)ation Ch 11 #7 R-Li is more reactive than R-MgX is. C–Li more polar than C–Mg C of R-Li more nu-philic [better Nu:] transmetalation [metal exchange] to less reactive [more stable] organometallic Coupling using Gilman reagent Ch 11 #8 coupling reaction (in organic chemistry) two hydrocarbon fragments are coupled (to form C–C) with the aid of a (transition) metal catalyst Gilman reagent coupling of R of R-X and R’ of Gilman reagent RX + R’2CuLi R–R’ mechanism? substitution of X with R’? not clear Ch 11 #9 R can be alkyl, aryl, or alkenyl [vinyl] which is not possible by R-Li or R-MgX Is R of Gilman why? they are SN(2). -

1 Structure, Properties, and Preparation of Boronic Acid Derivatives Overview of Their Reactions and Applications Dennis G
j1 1 Structure, Properties, and Preparation of Boronic Acid Derivatives Overview of Their Reactions and Applications Dennis G. Hall 1.1 Introduction and Historical Background Structurally, boronic acids are trivalent boron-containing organic compounds that possess one carbon-based substituent (i.e., a CÀB bond) and two hydroxyl groups to fill the remaining valences on the boron atom (Figure 1.1). With only six valence electrons and a consequent deficiency of two electrons, the sp2-hybridized boron atom possesses a vacant p-orbital. This low-energy orbital is orthogonal to the three substituents, which are oriented in a trigonal planar geometry. Unlike carbox- ylic acids, their carbon analogues, boronic acids, are not found in nature. These abiotic compounds are derived synthetically from primary sources of boron such as boric acid, which is made by the acidification of borax with carbon dioxide. Borate esters, one of the key precursors of boronic acid derivatives, are made by simple dehydration of boric acid with alcohols. The first preparation and isolation of a boronic acid was reported by Frankland in 1860 [1]. By treating diethylzinc with triethylborate, the highly air-sensitive triethylborane was obtained, and its slow oxidation in ambient air eventually provided ethylboronic acid. Boronic acids are the products of a twofold oxidation of boranes. Their stability to atmospheric oxidation is considerably superior to that of borinic acids, which result from the first oxidation of boranes. The product of a third oxidation of boranes, boric acid, is a very stable and relatively benign compound to humans (Section 1.2.2.3). Their unique properties and reactivity as mild organic Lewis acids, coupled with their stability and ease of handling, are what make boronic acids a particularly attractive class of synthetic intermediates. -

RSC Advances PAPER
View Article Online / Journal Homepage / Table of Contents for this issue RSC Advances Dynamic Article Links Cite this: RSC Advances, 2012, 2, 3968–3977 www.rsc.org/advances PAPER Palladium nanoshells coated with self-assembled monolayers and their catalytic properties Jun-Hyun Kim,*a Joon-Seo Park,b Hae-Won Chung,c Brett W. Bootea and T. Randall Lee*c Received 13th October 2011, Accepted 6th February 2012 DOI: 10.1039/c2ra00883a This report describes the preparation and characterization of palladium nanoshells protected with alkanethiol self-assembled monolayers (SAMs) and their application as efficient catalysts. Monodispersed silica core particles (y100 nm in diameter) were prepared and coated with a thin layer of palladium (y20 nm in thickness). Subsequently, the palladium nanoshells were treated with two separate alkanethiol adsorbates having different alkyl chain lengths: dodecanethiol (C12SH) and hexadecanethiol (C16SH). The optical properties, morphology, and chemical structure/composition of these nanoshells were thoroughly examined by ultraviolet-visible spectroscopy, field-emission scanning electron microscopy, transmission electron microscopy, energy-dispersive X-ray spectroscopy, X-ray photoelectron spectroscopy, and Fourier-transform infrared spectroscopy. Additional studies revealed that these SAM-coated palladium nanoshells possessed enhanced colloidal stability in nonpolar solvents and in the solid state. Further, palladium nanoshells modified with C16SH SAM coatings were employed in the Suzuki coupling of phenylboronic acid with iodobenzene in organic solvents. Notably, these SAM-coated nanoshells afforded a greater conversion yield than that of related bare palladium nanoshells. Downloaded on 19 February 2013 Introduction palladium nanoparticles as catalysts depend largely on their size, shape, surrounding medium, and dispersion ability in organic The controlled fabrication of nanoscale metallic particles offers the solvents, which facilitates their manipulation and incorporation 1–5 opportunity to develop novel catalysts. -
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Grignard Reaction: Synthesis of Triphenylmethanol
*NOTE: Grignard reactions are very moisture sensitive, so all the glassware in the reaction (excluding the work-up) should be dried in an oven with a temperature of > 100oC overnight. The following items require oven drying. They should be placed in a 150mL beaker, all labeled with a permanent marker. 1. 5mL conical vial (AKA: Distillation receiver). 2. Magnetic spin vane. 3. Claisen head. 4. Three Pasteur pipettes. 5. Two 1-dram vials (Caps EXCLUDED). 6. One 2-dram vial (Caps EXCLUDED). 7. Glass stirring rod 8. Adaptor (19/22.14/20) Grignard Reaction: Synthesis of Triphenylmethanol Pre-Lab: In the “equations” section, besides the main equations, also: 1) draw the equation for the production of the byproduct, Biphenyl. 2) what other byproduct might occur in the reaction? Why? In the “observation” section, draw data tables in the corresponding places, each with 2 columns -- one for “prediction” (by answering the following questions) and one for actual drops or observation. 1) How many drops of bromobenzene should you add? 2) How many drops of ether will you add to flask 2? 3) 100 µL is approximately how many drops? 4) What are the four signs of a chemical reaction? (Think back to Chem. 110) 5) How do the signs of a chemical reaction apply to this lab? The Grignard reaction is a useful synthetic procedure for forming new carbon- carbon bonds. This organometallic chemical reaction involves alkyl- or aryl-magnesium halides, known as Grignard 1 reagents. Grignard reagents are formed via the action of an alkyl or aryl halide on magnesium metal.