Hydrogen Segregation and Its Roles in Structural Stability and Metallization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
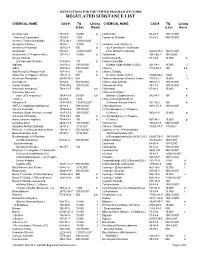
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Solid-State Structures of the Covalent Hydrides Germane and Stannane
Edinburgh Research Explorer Solid-state structures of the covalent hydrides germane and stannane Citation for published version: Maley, IJ, Brown, DH, Ibberson, RM & Pulham, CR 2008, 'Solid-state structures of the covalent hydrides germane and stannane', Acta Crystallographica Section B - Structural Science, vol. 64, no. Pt 3, pp. 312-7. https://doi.org/10.1107/S0108768108010379 Digital Object Identifier (DOI): 10.1107/S0108768108010379 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Acta Crystallographica Section B - Structural Science Publisher Rights Statement: Copyright © 2008 International Union of Crystallography; all rights reserved. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 electronic reprint Acta Crystallographica Section B Structural Science, Crystal Engineering and Materials ISSN 2052-5192 Solid-state structures of the covalent hydrides germane and stannane Iain J. Maley, Daniel H. Brown, Richard M. Ibberson and Colin R. Pulham Acta Cryst. (2008). B64, 312–317 Copyright c International Union of Crystallography Author(s) of this paper may load this reprint on their own web site or institutional repository provided that this cover page is retained. -

Acutely / Extremely Hazardous Waste List
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extemely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extemely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extemely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extemely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extemely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extemely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extemely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extemely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extemely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extemely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extemely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

United States Patent to 11, 4,018,606 Contois Et Al
United States Patent to 11, 4,018,606 Contois et al. 45 Apr. 19, 1977 (54) ORGANIC AZO PIGMENT SENSITIZERS FOR PHOTOCONDUCTIVE LAYERS R ph C 2 x (75) Inventors: Lawrence E. Contois; Joseph Y. R2 NEN-C Kaukeinen; Stephen Michel; Thomas C M. Plutchak, all of Rochester, N.Y. R R (73) Assignee: Eastman Kodak Company, wherein X consists of the atoms necessary to complete Rochester, N.Y. a naphthalene, anthracene, or OH (22 Filed: May 3, 1974 C 2 N. -C N N (21) Appl. No.: 466,658 =N R (52) U.S. Cl. ...................................... 96/1.7; 96/1.6 ring; (51) Int. Cl”.......................................... G03G 5/09 (58) Field of Search ........................... 96/1, 1.5, 1.6 R', R', and R are hydrogen, halogen, alkoxy, NO, alkyl, SOH or alkali metal salts thereof, (56) References Cited O O UNITED STATES PATENTS rCNH NO, and -CNH 3,384,632 5/1968 Solodar ................................ 96/1.6 3,622,341 8/1969 Lee ....................................... 96/1.6 and COOH or alkali metal salts thereof, and Rand R8 3,684,548 8/1972 Contois ............................... 96/1.6 can comprise the atoms necessary to complete a 3,775,105 l/1973 Kukla ................................... 96/1.6 phenyl, naphthyl or anthryl ring; and R is selected from FOREIGN PATENTS OR APPLICATIONS the group consisting of 1,370,197 10/1974 United Kingdom .................. 96/1.5 O O H il H Primary Examiner-David Klein 8-i-O-No, 8-N-O). or COOM Assistant Examiner-John L. Goodrow Attorney, Agent, or Firm-Arthur H. Rosenstein where M is alkyl, alkali or alkaline earth metal are useful as sensitizers for photoconductive compositions in electrophotographic processes. -

40 CFR Ch. I (7–1–14 Edition) Pt. 68, App. A
Pt. 68, App. A 40 CFR Ch. I (7–1–14 Edition) under subpart G of this part as required (j) Nothing in this section shall pre- by the final determination, and sub- clude, limit, or interfere in any way mits the revised RMP as required with the authority of EPA or the state under § 68.150. to exercise its enforcement, investiga- (i) The public shall have access to the tory, and information gathering au- preliminary determinations, responses, thorities concerning this part under and final determinations under this the Act. section in a manner consistent with § 68.210. APPENDIX A TO PART 68—TABLE OF TOXIC ENDPOINTS [As defined in § 68.22 of this part] Toxic end- CAS No. Chemical name point (mg/L) 107–02–8 ............. Acrolein [2-Propenal] ................................................................................................................ 0 .0011 107–13–1 ............. Acrylonitrile [2-Propenenitrile] ................................................................................................... 0.076 814–68–6 ............. Acrylyl chloride [2-Propenoyl chloride] ..................................................................................... 0 .00090 107–18–6 ............. Allyl alcohol [2-Propen-1-ol] ...................................................................................................... 0 .036 107–11–9 ............. Allylamine [2-Propen-1-amine] ................................................................................................. 0 .0032 7664–41–7 ........... Ammonia (anhydrous) ............................................................................................................. -

Stannane 2629
TRIMETHPLSTANNANE 2629 [CONTRIBUTION FROM THE CHEWCAL LABORATORY OF CLARK UNIVERSITY, I, 161 THE PREPARATION AND PROPERTIES OF TRIMETHYL- STANNANE BY CHARLESA. KRAUSAUD WILLARDTu'. GREER Recci\ed .4ugiist 11 1922 Introduction.-The hydrogen derivatives of the various elements decreasc in stability as we proceed from the elements of the halogen group to the elements of higher negative 1-alence,and as wc proceed from elements of lower to elements of higher atomic weight. A11 the elements of the halogen group give stable hydrides at ordinary temperatures. The hydrides of the higher member4 of the oxygen group have comparatively little stability, while in the case of the nitrogen group only nitrogen and phosphorus form comparatively stable normal hydrides. Of the fourth group of elements, carbon and silicon form stable hydrides, but the higher members form hydrides possessing little stability. It is only recently that the existence of hydrides o€ tin, lend and bismuth has been estab- lished.' ff one or more of the 111 drogens of a hydride is replaced by an alkyl or am1 group, the stability of the resulting compound is greatly increased over that of the hydride. Kearly all the common elements yield stable compounds of this type when all the hydrogens are substituted in this way. Tlie metallo-organic compounds of the elements of the fourth group exhibit unusual stability, corresponding somewhat to the exceptional stability of methane, the first member of the group. The tetra-alkyl tin compounds are, on the whole, very stable substances. It might be expected, therefore, that, in the case of tin if only a portion of the hydrogens mere wbstituted by alkyl groups, the resulting compound would exhibit a con- siclt~abledegree of stability. -

Environmental Protection Agency § 68.130
Environmental Protection Agency § 68.130 include, but are not limited to, phys- TABLE 1 TO § 68.130—LIST OF REGULATED ical and chemical properties of the sub- TOXIC SUBSTANCES AND THRESHOLD QUAN- stance, such as vapor pressure; mod- TITIES FOR ACCIDENTAL RELEASE PREVEN- eling results, including data and as- TION—Continued sumptions used and model documenta- [Alphabetical Order—77 Substances] tion; and historical accident data, cit- ing data sources. Threshold Chemical name CAS No. quantity Basis for (h) Within 18 months of receipt of a (lbs) listing petition, the Administrator shall pub- Ammonia (anhy- 7664–41–7 10,000 a, b lish in the FEDERAL REGISTER a notice drous). either denying the petition or granting Ammonia (conc 7664–41–7 20,000 a, b the petition and proposing a listing. 20% or greater). Arsenous tri- 7784–34–1 15,000 b § 68.125 Exemptions. chloride. Arsine ................. 7784–42–1 1,000 b Agricultural nutrients. Ammonia used Boron trichloride 10294–34–5 5,000 b as an agricultural nutrient, when held [Borane, by farmers, is exempt from all provi- trichloro-]. Boron trifluoride 7637–07–2 5,000 b sions of this part. [Borane, trifluoro-]. § 68.126 Exclusion. Boron trifluoride 353–42–4 15,000 b compound with Flammable Substances Used as Fuel or methyl ether Held for Sale as Fuel at Retail Facilities. (1:1) [Boron, A flammable substance listed in Tables trifluoro [oxybis 3 and 4 of § 68.130 is nevertheless ex- [metane]]-, T-4-. Bromine .............. 7726–95–6 10,000 a, b cluded from all provisions of this part Carbon disulfide 75–15–0 20,000 b when the substance is used as a fuel or Chlorine ............. -

High Pressure Stabilization and Emergent Forms of Pbh4
week ending PRL 107, 037002 (2011) PHYSICAL REVIEW LETTERS 15 JULY 2011 High Pressure Stabilization and Emergent Forms of PbH4 Patryk Zaleski-Ejgierd,1 Roald Hoffmann,2 and N. W. Ashcroft1 1Laboratory of Atomic and Solid State Physics and Cornell Center for Materials Research, Clark Hall, Cornell University, Ithaca, New York 14853-2501, USA 2Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, New York 14853-1301, USA (Received 17 October 2010; revised manuscript received 9 March 2011; published 11 July 2011; corrected 20 July 2011) A wide decomposition pressure range of 132 GPa is predicted for PbH4 above which it emerges in very different forms compared with its group-14 congeners. This triply Born-Oppenheimer system is a nonmolecular, three-dimensional, metallic alloy, despite a prominent layered structure. A significant number of enthalpically near-degenerate structures, with exceedingly small energy barriers for distortions, and characteristic instabilities in the phonon spectra suggest that even at very high pressures PbH4 may exhibit both metallic and liquidlike properties and sublattice or even full melting. DOI: 10.1103/PhysRevLett.107.037002 PACS numbers: 74.70.Ad, 74.62.Fj Among group-14 hydrides, the lead-hydrogen system is As starting points and guides in a structure search, we special, for it clearly combines one of the heaviest (Pb) used a number of geometries previously reported for group- elements with the lightest (H). The attendant mass ratio 14 tetrahydrides, EH4, as well as other structures in which (207:1) immediately implies a distinct separation of time lead is two- to 16-fold coordinated by hydrogen, and with scales for the ensuing dynamics, in very much the way that up to Z ¼ 4 formula units per cell. -

CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011
CSAT Top-Screen Questions January 2009 Version 2.8 CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011 Change Log .........................................................................................................3 CVI Authorizing Statements...............................................................................4 General ................................................................................................................6 Facility Description.................................................................................................................... 7 Facility Regulatory Mandates ................................................................................................... 7 EPA RMP Facility Identifier....................................................................................................... 9 Refinery Capacity....................................................................................................................... 9 Refinery Market Share ............................................................................................................. 10 Airport Fuels Supplier ............................................................................................................. 11 Military Installation Supplier................................................................................................... 11 Liquefied Natural Gas (LNG) Capacity................................................................................... 12 Liquefied Natural Gas Exclusion -

Select Reactions of Organoboranes and Organostannanes
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2012 Select Reactions of Organoboranes and Organostannanes David W. Blevins [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Organic Chemistry Commons Recommended Citation Blevins, David W., "Select Reactions of Organoboranes and Organostannanes. " PhD diss., University of Tennessee, 2012. https://trace.tennessee.edu/utk_graddiss/1412 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by David W. Blevins entitled "Select Reactions of Organoboranes and Organostannanes." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. George W. Kabalka, Major Professor We have read this dissertation and recommend its acceptance: Shane Foister, Craig Barnes, Kimberly Gwinn Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Select Reactions of Organoboranes and Organostannanes A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville David W. Blevins August 2012 DEDICATION I dedicate this work to Lisa, Preston, and Clarissa Blevins. ii ACKNOWLEDGEMENTS I would like to thank Dr. -

EPA's Hazardous Waste Listing
Hazardous Waste Listings A User-Friendly Reference Document September 2012 Table of Contents Introduction ..................................................................................................................................... 3 Overview of the Hazardous Waste Identification Process .............................................................. 5 Lists of Hazardous Wastes .............................................................................................................. 5 Summary Chart ............................................................................................................................... 8 General Hazardous Waste Listing Resources ................................................................................. 9 § 261.11 Criteria for listing hazardous waste. .............................................................................. 11 Subpart D-List of Hazardous Wastes ............................................................................................ 12 § 261.31 Hazardous wastes from non-specific sources. ............................................................... 13 Spent solvent wastes (F001 – F005) ......................................................................................... 13 Wastes from electroplating and other metal finishing operations (F006 - F012, and F019) ... 18 Dioxin bearing wastes (F020 - F023, and F026 – F028) .......................................................... 22 Wastes from production of certain chlorinated aliphatic hydrocarbons (F024 -

Hydrides at High Pressures; an Evaluation of "Chemical Precompression" on the Way to Metallic Hydrogen
Hydrides at High Pressures; An Evaluation of "Chemical Precompression" on the way to Metallic Hydrogen by Paulina Gonzalez Morelos This thesis/dissertation document has been electronically approved by the following individuals: Hoffmann,Roald (Chairperson) Lee,Stephen (Minor Member) Chan,Garnet (Minor Member) HYDRIDES AT HIGH PRESSURES; AN EVALUATION OF “CHEMICAL PRECOMPRESSION” ON THE WAY TO METALLIC HYDROGEN A Dissertation Presented to the Faculty of the Graduate School of Cornell University in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Paulina Gonzalez Morelos August 2010 c 2010 Paulina Gonzalez Morelos ALL RIGHTS RESERVED HYDRIDES AT HIGH PRESSURES; AN EVALUATION OF “CHEMICAL PRECOMPRESSION” ON THE WAY TO METALLIC HYDROGEN Paulina Gonzalez Morelos, Ph.D. Cornell University 2010 In this dissertation, we present a theoretical study of compounds rich in hydro- gen, focusing on their crystal structure and potential metallization as pressure is applied. Chapter 1 is an introduction to metallic hydrogen; it reviews previous stud- ies and suggests a new way of analyzing the geometric changes that ensue as pressure is applied. Chapter 2 describes the search for structures for SnH4, us- ing chemical intuition and random search algorithms. Chapter 3 deals with the general problem of segregation which arises as SnH4, a thermodynamically metastable (positive heat of formation from Sn and 2H2) but kinetically persis- tent compound, is compressed. Chapter 3 is a complement to Chapter 2, as layered structures arise naturally in the low pressure regime, prompting us to explore the subject. Chapter 4 begins an ambitious project which strives to understand the elec- tronic structure and properties of metallic hydrides of the form MHx, where M= Si, Sn and W, and x = 4, 6, 8, 10, 12.