Studies of the Hydrides of the Heavier Group 14 Elements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
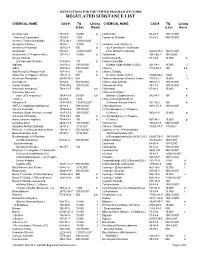
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Use of Solvents for Pahs Extraction and Enhancement of the Pahs Bioremediation in Coal- Tar-Contaminated Soils Pak-Hing Lee Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2000 Use of solvents for PAHs extraction and enhancement of the PAHs bioremediation in coal- tar-contaminated soils Pak-Hing Lee Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Environmental Engineering Commons Recommended Citation Lee, Pak-Hing, "Use of solvents for PAHs extraction and enhancement of the PAHs bioremediation in coal-tar-contaminated soils " (2000). Retrospective Theses and Dissertations. 13912. https://lib.dr.iastate.edu/rtd/13912 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter fece, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quaiity of the copy submitted. Broken or indistinct print colored or poor quality illustrations and photographs, print bleedthrough, substeindard margins, and improper alignment can adversely affect reproduction. In the unlilcely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -

Determination of Parent and Alkylated Polycyclic Aromatic Hydrocarbons (Pahs) in Biota and Sediment
ICES TECHNIQUES IN MARINE ENVIRONMENTAL SCIENCES NO. 45 NOVEMBER 2009 Determination of parent and alkylated polycyclic aromatic hydrocarbons (PAHs) in biota and sediment L. WEBSTER • J. TRONCZYNSKI P. KORYTAR • K. BOOIJ • R. LAW International Council for the Exploration of the Sea Conseil International pour l’Exploration de la Mer H. C. Andersens Boulevard 44 – 46 DK‐1553 Copenhagen V Denmark Telephone (+45) 33 38 67 00 Telefax (+45) 33 93 42 15 www.ices.dk [email protected] Our cover photo was taken by N. Penny Holliday aboard the RRS “Discovery” in rough seas in the Rockall Trough. Recommended format for purposes of citation: Webster, L., Tronczynski, J., Korytar, P., Booij, K., and Law, R. 2010. Determination of parent and alkylated polycyclic aromatic hydrocarbons (PAHs) in biota and sediment. ICES Techniques in Marine Environmental Sciences. No. 45. 26 pp. Series Editor: Paul D. Keizer For permission to reproduce material from this publication, please apply directly to the General Secretary. Correspondence concerning the details of any method or procedure should be directed to the author(s). This series presents detailed descriptions of methods and procedures relating to chemical and biological measurements in the marine environment. Most techniques described have been selected for documentation based on performance in ICES or other intercalibration or intercomparison exercises: they have been carefully evaluated and shown to yield good results when correctly applied. They have also been subject to review by relevant ICES working groups, but this is not to be construed as constituting official recommendation by the Council. ISBN 978-87-7482-074-1 978-87-7482-297-4 https://doi.org/10.17895/ices.pub.5070 ISSN 0903 – 2606 2707-6997 © 2010 International Council for the Exploration of the Sea ICES Techniques in Marine Environmental Sciences No. -

An Investigation of the Crystal Growth of Heavy Sulfides in Supercritical
AN ABSTRACT OF THE THESIS OF LEROY CRAWFORD LEWIS for the Ph. D. (Name) (Degree) in CHEMISTRY presented on (Major) (Date) Title: AN INVESTIGATION OF THE CRYSTAL GROWTH OF HEAVY SULFIDES IN SUPERCRITICAL HYDROGEN SULFIDE Abstract approved Redacted for privacy Dr. WilliarriIJ. Fredericks Solubility studies on the heavy metal sulfides in liquid hydrogen sulfide at room temperature were carried out using the isopiestic method. The results were compared with earlier work and with a theoretical result based on Raoult's Law. A relative order for the solubilities of sulfur and the sulfides of tin, lead, mercury, iron, zinc, antimony, arsenic, silver, and cadmium was determined and found to agree with the theoretical result. Hydrogen sulfide is a strong enough oxidizing agent to oxidize stannous sulfide to stannic sulfide in neutral or basic solution (with triethylamine added). In basic solution antimony trisulfide is oxi- dized to antimony pentasulfide. In basic solution cadmium sulfide apparently forms a bisulfide complex in which three moles of bisul- fide ion are bonded to one mole of cadmium sulfide. Measurements were made extending the range over which the volumetric properties of hydrogen sulfide have been investigated to 220 °C and 2000 atm. A virial expression in density was used to represent the data. Good agreement, over the entire range investi- gated, between the virial expressions, earlier work, and the theorem of corresponding states was found. Electrical measurements were made on supercritical hydro- gen sulfide over the density range of 10 -24 moles per liter and at temperatures from the critical temperature to 220 °C. Dielectric constant measurements were represented by a dielectric virial ex- pression. -

Supporting Information of Polycyclic Aromatic Hydrocarbons (Pahs) In
Supporting Information of Polycyclic aromatic hydrocarbons (PAHs) in aerosols over the central Himalayas along two south-north transects Peng Fei Chen1,5, Chao Liu Li1, Shi Chang Kang2,3*, Maheswar Rupakheti4, Arnico K Panday6, Fang Ping Yan2,5, Quan Lian Li2, Qiang Gong Zhang1,3, Jun Ming Guo1,5, Dipesh Rupakheti1,5, Wei Luo7 1Key Laboratory of Tibetan Environment Changes and Land Surface Processes, Institute of Tibetan Plateau Research, Chinese Academy of Sciences, Beijing 100101, China 2State Key Laboratory of Cryospheric Science, Cold and Arid Regions Environmental and Engineering Research Institute, Lanzhou 730000, China 3Center for Excellence in Tibetan Plateau Earth Sciences, Chinese Academy of Sciences, Beijing 100085, China; 4Institute for Advanced Sustainability Studies, Potsdam 14467, Germany 5University of Chinese Academy of Sciences, Beijing 100039, China 6International Centre for Integrated Mountain Development, Kathmandu, Nepal 7State Key Lab of Urban and Regional Ecology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China Correspondence to: S. Kang ([email protected]) Text SI-1 Sample extraction and analysis Text SI-2 Quality control Table SI-1 Percentage contribution (%) of each species to total PAHs in the atmosphere over the Himalayas. Text SI-1 Sample extraction and analysis A quarter of each filter was cut into pieces, placed into a glass tube, and immersed in 20 mL of dichloromethane (DCM) and n-hexane (1:1). The extraction was performed by sonication twice for 30 min at 27 °C. Every single sample was spiked with deuterated PAHs (naphthalene-d8, acenaphthene-d10, phenanthrene-d10, chrysene-d12, and perylene-d12) as recovery surrogates. -

Safety Data Sheet
SAFETY DATA SHEET Revision Date 27-February-2020 Revision Number 2 1. Identification Product Name Potassium hydride, 30% w/w in mineral oil Cat No. : L13266 Synonyms No information available Recommended Use Laboratory chemicals. Uses advised against Food, drug, pesticide or biocidal product use. Details of the supplier of the safety data sheet Company Importer/Distributor Manufacturer Fisher Scientific Alfa Aesar 112 Colonnade Road, Thermo Fisher Scientific Chemicals, Inc. Ottawa, ON K2E 7L6, 30 Bond Street, Ward Hill, MA 01835-8099 Canada Tel: 800-343-0660 Fax: 800-322-4757 Tel: 1-800-234-7437 Email: [email protected] www.alfa.com Emergency Telephone Number During normal business hours (Monday-Friday, 8am-7pm EST), call (800) 343-0660. After normal business hours, call Carechem 24 at (800) 579-7421. 2. Hazard(s) identification Classification WHMIS 2015 Classification Classified as hazardous under the Hazardous Products Regulations (SOR/2015-17) Substances/mixtures which, in contact with water, emit Category 1 flammable gases Skin Corrosion/Irritation Category 1 B Serious Eye Damage/Eye Irritation Category 1 Specific target organ toxicity (single exposure) Category 3 Target Organs - Respiratory system. Aspiration Toxicity Category 1 Physical Hazards Not Otherwise Classified Category 1 Reacts violently with water Label Elements Signal Word Danger Hazard Statements In contact with water releases flammable gases which may ignite spontaneously ______________________________________________________________________________________________ Page 1 / 7 Potassium hydride, 30% w/w in mineral oil Revision Date 27-February-2020 ______________________________________________________________________________________________ May be fatal if swallowed and enters airways Causes severe skin burns and eye damage May cause respiratory irritation Reacts violently with water Precautionary Statements Manufacturer Alfa Aesar Thermo Fisher Scientific Chemicals, Inc. -

Page 1 of 26 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 26 RSC Advances The ternary amide KLi 3(NH 2)4: an important intermediate in the potassium compounds-added Li –N–H systems Bao-Xia Dong, Liang Song, Jun Ge, Yun-Lei Teng*, Shi-Yang Zhang College of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, P. R. China. In this paper, the KH-added LiH–NH 3, KH-added LiH–LiNH 2, KH-added LiNH 2, and KNH 2-added LiNH 2 systems were systematically investigated. It was found that the ternary amide KLi 3(NH 2)4 was an important intermediate that was inclined to be formed in the dehydrogenation and hydrogenation processes of the potassium compounds-added Li–N–H system. -

United States Patent Office 2,807,613
United States Patent Office 2,807,613 Patented Sept. 24, 1957 s s 2 2,807,613 nol, where it may exist in the form of a hemiformal, or PREPARATION OF 6-METHYL-6-PHENYLTETRA of a revertible polymer. Usually formaldehyde is used in HYDRO-1,3-OXAZINES excess based on molar proportions referred to the a Claude 5. Schmide, Moorestown, and Richard C. Maas 5 methylstyrene, proportions from about 1.5:1 to 5:1 being field, Haddon afield, N. J., assignors to Rohm & Hiaas practical. Of course, with less than a 2:1 proportion Company, Philadelphia, Pa., a corporatica of Delaware unreacted starting materials may be present in the react ing mixture, Preferred proportions are from 2:1 to 4:1. No Drawing. Application April 3, 1955, Ammonia may be supplied as a gas or as an aqueous Serial No. 577,944 solution or in the form of ammonium chloride or bromide. 7 Claims. (C. 260-244) 0 Of course, if ammonia or ammonium hydroxide is used, This invention deals with a method for improving yields it will react with the hydrochloric or hydrobromic acid of 6-methyl-6-phenyltetrahydro-1,3-oxazines when made which is added as catalyst. The same final result is ob from an O-methylstyrene, formaldehyde, and ammonia. tained by use of the preformed ammonium halide, which In United States Patent 2,647,117, there is described 5 Supplies both the ammonia and the catalyst. The amount the reaction of olefins, including c-methylstyrene, with of ammonia or ammonium compound is usually at least ammonia and formaldehyde in the presence of hydrogen equivalent to the c-methylstyrene and may be in consid -chloride as a catalyst. -

Effect of Epoxide Content on the Vulcanizate Structure of Silica-Filled Epoxidized Natural Rubber (ENR) Compounds
polymers Article Effect of Epoxide Content on the Vulcanizate Structure of Silica-Filled Epoxidized Natural Rubber (ENR) Compounds Gyeongchan Ryu 1, Donghyuk Kim 1, Sanghoon Song 1, Kiwon Hwang 1, Byungkyu Ahn 2 and Wonho Kim 1,* 1 School of Chemical Engineering, Pusan National University, Busan 46241, Korea; [email protected] (G.R.); [email protected] (D.K.); [email protected] (S.S.); [email protected] (K.H.) 2 Hankook Tire & Technology Co., Ltd., R&D Center, 50 Yuseong-daero 935 beon-gil, Yuseong-gu, Daejeon 34127, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-51-510-3190 Abstract: The demand for truck–bus radial (TBR) tires with enhanced fuel efficiency has grown in recent years. Many studies have investigated silica-filled natural rubber (NR) compounds to address these needs. However, silica-filled compounds offer inferior abrasion resistance compared to carbon black-filled compounds. Further, the use of NR as a base rubber can hinder silanization and coupling reactions due to interference by proteins and lipids. Improved silica dispersion be achieved without the use of a silane coupling agent by introducing epoxide groups to NR, which serve as silica-affinitive functional groups. Furthermore, the coupling reaction can be promoted by facilitating chemical interaction between the hydroxyl group of silica and the added epoxide groups. Thus, this study evaluated the properties of commercialized NR, ENR-25, and ENR-50 compounds with or without an added silane coupling agent, and the filler–rubber interaction was quantitatively calculated using vulcanizate structure analysis. The increased epoxide content, when the silane coupling agent was not used, improved silica dispersion, abrasion resistance, fuel efficiency, and wet Citation: Ryu, G.; Kim, D.; Song, S.; grip. -

Organic Chemistry Name Formula Isomers Methane CH 1 Ethane C H
Organic Chemistry Organic chemistry is the chemistry of carbon. The simplest carbon molecules are compounds of just carbon and hydrogen, hydrocarbons. We name the compounds based on the length of the longest carbon chain. We then add prefixes and suffixes to describe the types of bonds and any add-ons the molecule has. When the molecule has just single bonds we use the -ane suffix. Name Formula Isomers Methane CH4 1 Ethane C2H6 1 Propane C3H8 1 Butane C4H10 2 Pentane C5H12 3 Hexane C6H14 5 Heptane C7H16 9 Octane C8H18 18 Nonane C9H20 35 Decane C10H22 75 Isomers are compounds that have the same formula but different bonding. isobutane n-butane 1 Naming Alkanes Hydrocarbons are always named based on the longest carbon chain. When an alkane is a substituent group they are named using the -yl ending instead of the -ane ending. So, -CH3 would be a methyl group. The substituent groups are named by numbering the carbons on the longest chain so that the first branching gets the lowest number possible. The substituents are listed alphabetically with out regard to the number prefixes that might be used. 3-methylhexane 1 2 3 4 5 6 6 5 4 3 2 1 Alkenes and Alkynes When a hydrocarbon has a double bond we replace the -ane ending with -ene. When the hydrocarbon has more than three carbon the position of the double bond must be specified with a number. 1-butene 2-butene Hydrocarbons with triple bonds are named basically the same, we replace the -ane ending with -yne. -

Stoichiometric and Catalytic Reactions Involving Si-H Bond Activations by Rh and Ir Complexes Containing a Pyridylindolide Ligand Dmitry Karshtedt,†,‡ Alexis T
Organometallics 2006, 25, 4471-4482 4471 Stoichiometric and Catalytic Reactions Involving Si-H Bond Activations by Rh and Ir Complexes Containing a Pyridylindolide Ligand Dmitry Karshtedt,†,‡ Alexis T. Bell,*,§,‡ and T. Don Tilley*,†,‡ Departments of Chemistry and Chemical Engineering, UniVersity of California, Berkeley, Berkeley, California 94720, and Chemical Sciences DiVision, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720 ReceiVed June 5, 2006 New rhodium and iridium complexes containing the bidentate, monoanionic 2-(2′-pyridyl)indolide (PyInd) ligand were prepared. The bis(ethylene) complex (PyInd)Rh(C2H4)2 (3) reacted with triethylsilane at 60 °C to produce the 16-electron Rh(V) bis(silyl)dihydride complex (PyInd)RhH2(SiEt3)2 (4). Both 3 and 4 catalyzed the chloride transfer reaction of chlorobenzene and Et3SiH to give Et3SiCl and benzene i in the absence of base. In the presence of LiN Pr2, catalytic C-Si coupling was observed, to produce Et3SiPh. Analogous Ir complexes of the type (PyInd)IrH2(SiR3)2, where R3 ) Et3 (6a), Me2Ph (6b), Ph2Me (6c), or Ph3 (6d), were not catalytically active in this chloride transfer chemistry. The molecular structure of 6c, determined by X-ray crystallography, may be described as having a highly unusual bicapped tetrahedral geometry. DFT calculations indicate that this distortion is associated with the d4 electron count and the presence of highly trans-influencing silyl ligands. The reaction of 6a with PMe3 (5 equiv) produced (PyInd)IrH(SiEt3)(PMe3)2 (7), while the reaction with PPh3 (1 equiv) generated a mixture of isomers that was converted to (PyInd)IrH(SiEt3)(PPh3)(8) upon heating in benzene at 80 °C.