Comparison of Different Functional Prediction Scores Using a Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -
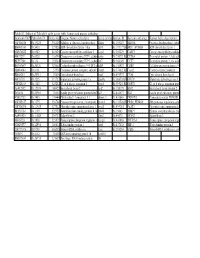
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

A Rare Variant in MCF2L Identified Using Exclusion Linkage in A
European Journal of Human Genetics (2016) 24, 86–91 & 2016 Macmillan Publishers Limited All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE A rare variant in MCF2L identified using exclusion linkage in a pedigree with premature atherosclerosis Stephanie Maiwald1,7, Mahdi M Motazacker1,7,8, Julian C van Capelleveen1, Suthesh Sivapalaratnam1, Allard C van der Wal2, Chris van der Loos2, John JP Kastelein1, Willem H Ouwehand3,4, G Kees Hovingh1, Mieke D Trip1,5, Jaap D van Buul6 and Geesje M Dallinga-Thie*,1 Cardiovascular disease (CVD) is a major cause of death in Western societies. CVD risk is largely genetically determined. The molecular pathology is, however, not elucidated in a large number of families suffering from CVD. We applied exclusion linkage analysis and next-generation sequencing to elucidate the molecular defect underlying premature CVD in a small pedigree, comprising two generations of which six members suffered from premature CVD. A total of three variants showed co-segregation with the disease status in the family. Two of these variants were excluded from further analysis based on the prevalence in replication cohorts, whereas a non-synonymous variant in MCF.2 Cell Line Derived Transforming Sequence-like protein (MCF2L, c.2066A4G; p.(Asp689Gly); NM_001112732.1), located in the DH domain, was only present in the studied family. MCF2L is a guanine exchange factor that potentially links pathways that signal through Rac1 and RhoA. Indeed, in HeLa cells, MCF2L689Gly failed to activate Rac1 as well as RhoA, resulting in impaired stress fiber formation. Moreover, MCF2L protein was found in human atherosclerotic lesions but not in healthy tissue segments. -

Novel Targets of Apparently Idiopathic Male Infertility
International Journal of Molecular Sciences Review Molecular Biology of Spermatogenesis: Novel Targets of Apparently Idiopathic Male Infertility Rossella Cannarella * , Rosita A. Condorelli , Laura M. Mongioì, Sandro La Vignera * and Aldo E. Calogero Department of Clinical and Experimental Medicine, University of Catania, 95123 Catania, Italy; [email protected] (R.A.C.); [email protected] (L.M.M.); [email protected] (A.E.C.) * Correspondence: [email protected] (R.C.); [email protected] (S.L.V.) Received: 8 February 2020; Accepted: 2 March 2020; Published: 3 March 2020 Abstract: Male infertility affects half of infertile couples and, currently, a relevant percentage of cases of male infertility is considered as idiopathic. Although the male contribution to human fertilization has traditionally been restricted to sperm DNA, current evidence suggest that a relevant number of sperm transcripts and proteins are involved in acrosome reactions, sperm-oocyte fusion and, once released into the oocyte, embryo growth and development. The aim of this review is to provide updated and comprehensive insight into the molecular biology of spermatogenesis, including evidence on spermatogenetic failure and underlining the role of the sperm-carried molecular factors involved in oocyte fertilization and embryo growth. This represents the first step in the identification of new possible diagnostic and, possibly, therapeutic markers in the field of apparently idiopathic male infertility. Keywords: spermatogenetic failure; embryo growth; male infertility; spermatogenesis; recurrent pregnancy loss; sperm proteome; DNA fragmentation; sperm transcriptome 1. Introduction Infertility is a widespread condition in industrialized countries, affecting up to 15% of couples of childbearing age [1]. It is defined as the inability to achieve conception after 1–2 years of unprotected sexual intercourse [2]. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Bicbioec: Biclustering in Biomarker Identification for ESCC
BicBioEC: biclustering in biomarker identification for ESCC P. Kakati, D. K. Bhattacharyya & J. K. Kalita Network Modeling Analysis in Health Informatics and Bioinformatics ISSN 2192-6662 Volume 8 Number 1 Netw Model Anal Health Inform Bioinforma (2019) 8:1-21 DOI 10.1007/s13721-019-0200-x 1 23 Your article is protected by copyright and all rights are held exclusively by Springer-Verlag GmbH Austria, part of Springer Nature. This e- offprint is for personal use only and shall not be self-archived in electronic repositories. If you wish to self-archive your article, please use the accepted manuscript version for posting on your own website. You may further deposit the accepted manuscript version in any repository, provided it is only made publicly available 12 months after official publication or later and provided acknowledgement is given to the original source of publication and a link is inserted to the published article on Springer's website. The link must be accompanied by the following text: "The final publication is available at link.springer.com”. 1 23 Author's personal copy Network Modeling Analysis in Health Informatics and Bioinformatics (2019) 8:19 https://doi.org/10.1007/s13721-019-0200-x ORIGINAL ARTICLE BicBioEC: biclustering in biomarker identifcation for ESCC P. Kakati1 · D. K. Bhattacharyya1 · J. K. Kalita2 Received: 23 November 2018 / Revised: 26 June 2019 / Accepted: 21 July 2019 © Springer-Verlag GmbH Austria, part of Springer Nature 2019 Abstract Analysis of gene expression patterns enables identifcation of signifcant genes related to a specifc disease. We analyze gene expression data for esophageal squamous cell carcinoma (ESCC) using biclustering, gene–gene network topology and pathways to identify signifcant biomarkers. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Dysfunctional Nitric Oxide Signalling Increases Risk of Myocardial Infarction
LETTER doi:10.1038/nature12722 Dysfunctional nitric oxide signalling increases risk of myocardial infarction Jeanette Erdmann1,2*, Klaus Stark3,4*, Ulrike B. Esslinger3,5*, Philipp Moritz Rumpf6,7*, Doris Koesling8, Cor de Wit2,9, Frank J. Kaiser2,10, Diana Braunholz10, Anja Medack1, Marcus Fischer3, Martina E. Zimmermann3, Stephanie Tennstedt1, Elisabeth Graf11,12, Sebastian Eck11,12, Zouhair Aherrahrou1,2, Janja Nahrstaedt1, Christina Willenborg1,2, Petra Bruse1, Ingrid Brænne1, Markus M. No¨then13,14, Per Hofmann13,15, Peter S. Braund16,17, Evanthia Mergia8, Wibke Reinhard6,7, Christof Burgdorf6, Stefan Schreiber18, Anthony J. Balmforth19, Alistair S. Hall20, Lars Bertram21, Elisabeth Steinhagen-Thiessen22, Shu-Chen Li23,24, Winfried Ma¨rz25,26,27, Muredach Reilly28, Sekar Kathiresan29,30,31, Ruth McPherson32, Ulrich Walter33,34, CARDIoGRAM{, Jurg Ott35,36, Nilesh J. Samani16,17, Tim M. Strom11,12, Thomas Meitinger6,11,12, Christian Hengstenberg6,7 & Heribert Schunkert6,7 Myocardialinfarction,a leading cause of death in the Western world1, may provide a new therapeutic target for reducing the risk of myo- usually occurs when the fibrous cap overlying an atherosclerotic pla- cardial infarction. que in a coronary artery ruptures. The resulting exposure of blood The starting point of the present study (work flow in Supplementary to the atherosclerotic material then triggers thrombus formation, Fig. 1) was the identification of a family with 32 members diagnosed which occludes the artery2. The importance of genetic predisposi- with coronary artery disease (CAD), of whom 22 had early onset of the tion to coronary artery disease and myocardial infarction is best disease (#60 years of age). Given the high mortality rates of myocar- documented by the predictive value of a positive family history3. -

Network-Assisted Analysis of Primary Sjögren's Syndrome GWAS Data In
Network-assisted analysis of primary Sjögren’s syndrome GWAS data in Han Chinese Kechi Fang, Kunlin Zhang, Jing Wang* Address: Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China. Email: Kechi Fang – [email protected]; Kunlin Zhang – [email protected]; Jing Wang – [email protected] *Corresponding author 1 Supplementary materials Page 3 – Page 5: Supplementary Figure S1. The direct network formed by the module genes from DAPPLE. Page 6: Supplementary Figure S2. Transcript expression heatmap. Page 7: Supplementary Figure S3. Transcript enrichment heatmap. Page 8: Supplementary Figure S4. Workflow of network-assisted analysis of pSS GWAS data to identify candidate genes. Page 9 – Page 734: Supplementary Table S1. A full list of PPI pairs involved in the node-weighted pSS interactome. Page 735 – Page 737: Supplementary Table S2. Detailed information about module genes and sigMHC-genes. Page 738: Supplementary Table S3. GO terms enriched by module genes. 2 NFKBIE CFLAR NFKB1 STAT4 JUN HSF1 CCDC90B SUMO2 STAT1 PAFAH1B3 NMI GTF2I 2e−04 CDKN2C LAMA4 8e−04 HDAC1 EED 0.002 WWOX PSMD7 0.008 TP53 PSMA1 HR 0.02 RPA1 0.08 UBC ARID3A PTTG1 0.2 TSC22D4 ERH NIF3L1 0.4 MAD2L1 DMRTB1 1 ERBB4 PRMT2 FXR2 MBL2 CBS UHRF2 PCNP VTA1 3 DNMT3B DNMT1 RBBP4 DNMT3A RFC3 DDB1 THRA CBX5 EED NR2F2 RAD9A HUS1 RFC4 DDB2 HDAC2 HCFC1 CDC45L PPP1CA MLLSMARCA2 PGR SP3 EZH2 CSNK2B HIST1H4C HIST1H4F HNRNPUL1 HR HIST4H4 TAF1C HIST1H4A ENSG00000206300 APEX1 TFDP1 RHOA ENSG00000206406 RPF2 E2F4 HIST1H4IHIST1H4B HIST1H4D -
Supplementary Information Patients and Sample Preparation
Supplementary Information Transcriptome characterization by RNA sequencing identifies two major molecular subgroups with clinical relevance in chronic lymphocytic leukemia Patients and Sample Preparation RNA-sequencing studies were performed in CLL samples from 98 patients, 41 had IGHV-unmutated and 54 IGHV-mutated genes (<98% identity), and in 3 the IGHV mutational status could not be determined. The complete clinical and biological data at the moment of sampling and before any treatment were available in 91 of these patients (Table S2). One hundred and twenty four additional CLL patients sequentially included in the International Cancer Genome Consortium (ICGC) CLL project constituted a validation series and were studied by microarray expression profile. The complete clinical and biological data at the moment of sampling and before any treatment were available in 110 of these patients (Table S2). All patients gave informed consent for their participation in the study following the ICGC guidelines. The tumor samples used for RNA-sequencing were obtained from fresh or cryopreserved mononuclear cells. To purify the CLL fraction, samples were incubated with a cocktail of magnetically-labelled antibodies directed against T cells, NK cells, monocytes and granulocytes (CD2, CD3, CD11b, CD14, CD15 and CD56), adjusted to the percentage of each contaminating population (AutoMACS, Miltenyi Biotec). The degree of contamination by non-CLL cells in the CLL fraction was assessed by immunophenotype and flow cytometry and was lower than 5%. Normal samples were obtained from buffy coats from healthy adult donors with an average age of 54 years (ranging from 45 to 61, Table S5). After Ficoll-Isopaque density centrifugation CD19+ B cells were isolated by positive magnetic cell separation by using AutoMACS system (Milteny Biotec, Auburn, CA). -
Screening of Underlying Genetic Biomarkers for Ankylosing Spondylitis
MOLECULAR MEDICINE REPORTS 19: 5263-5274, 2019 Screening of underlying genetic biomarkers for ankylosing spondylitis XUTAO FAN1, BAO QI1, LONGFEI MA2 and FENGYU MA3 1Department of Spine Surgery, Affiliated Hospital of Jining Medical University, Jining, Shandong 272029; 2Graduate School of Jining Medical University, Jining, Shandong 272067; 3Department of Spine Surgery, People's Hospital of Rizhao, Rizhao, Shandong 276800, P.R. China Received August 6, 2018; Accepted March 6, 2019 DOI: 10.3892/mmr.2019.10188 Abstract. Genetic biomarkers for the diagnosis of anky- Introduction losing spondylitis (AS) remain unreported except for human leukocyte antigen B27 (HLA‑B27). Therefore, the aim of Ankylosing spondylitis (AS) is a common inflammatory rheu- the present study was to screen the differentially expressed matic disease, with an estimated prevalence (per 10,000) of genes (DEGs), and those that also possess differential single 23.8 in Europe, 16.7 in Asia, 31.9 in North America, 10.2 in nucleotide polymorphism (SNP) loci in the whole blood of Latin America and 7.4 in Africa (1). AS mainly affects the AS patients compared with healthy controls by integrating spine and sacroiliac joints in the pelvis to cause low back pain, two mRNA expression profiles (GSE73754 and GSE25101) stiffness and functional disability, which seriously influence and SNP microarray data (GSE39428) collected from the the quality of life of patients and impose a heavy economic Gene Expression Omnibus (GEO). Using the t-test, 1,056 burden on both family and society (2). Therefore, there is a and 1,073 DEGs were identified in the GSE73754 and need for the timely diagnosis and effective treatment of AS.