Bicbioec: Biclustering in Biomarker Identification for ESCC
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

Transcriptional Regulation of RKIP in Prostate Cancer Progression
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Sciences Transcriptional Regulation of RKIP in Prostate Cancer Progression Submitted by: Sandra Marie Beach In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Sciences Examination Committee Major Advisor: Kam Yeung, Ph.D. Academic William Maltese, Ph.D. Advisory Committee: Sonia Najjar, Ph.D. Han-Fei Ding, M.D., Ph.D. Manohar Ratnam, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: May 16, 2007 Transcriptional Regulation of RKIP in Prostate Cancer Progression Sandra Beach University of Toledo ACKNOWLDEGMENTS I thank my major advisor, Dr. Kam Yeung, for the opportunity to pursue my degree in his laboratory. I am also indebted to my advisory committee members past and present, Drs. Sonia Najjar, Han-Fei Ding, Manohar Ratnam, James Trempe, and Douglas Pittman for generously and judiciously guiding my studies and sharing reagents and equipment. I owe extended thanks to Dr. William Maltese as a committee member and chairman of my department for supporting my degree progress. The entire Department of Biochemistry and Cancer Biology has been most kind and helpful to me. Drs. Roy Collaco and Hong-Juan Cui have shared their excellent technical and practical advice with me throughout my studies. I thank members of the Yeung laboratory, Dr. Sungdae Park, Hui Hui Tang, Miranda Yeung for their support and collegiality. The data mining studies herein would not have been possible without the helpful advice of Dr. Robert Trumbly. I am also grateful for the exceptional assistance and shared microarray data of Dr. -

Epigenome-Wide Exploratory Study of Monozygotic Twins Suggests Differentially Methylated Regions to Associate with Hand Grip Strength
Biogerontology (2019) 20:627–647 https://doi.org/10.1007/s10522-019-09818-1 (0123456789().,-volV)( 0123456789().,-volV) RESEARCH ARTICLE Epigenome-wide exploratory study of monozygotic twins suggests differentially methylated regions to associate with hand grip strength Mette Soerensen . Weilong Li . Birgit Debrabant . Marianne Nygaard . Jonas Mengel-From . Morten Frost . Kaare Christensen . Lene Christiansen . Qihua Tan Received: 15 April 2019 / Accepted: 24 June 2019 / Published online: 28 June 2019 Ó The Author(s) 2019 Abstract Hand grip strength is a measure of mus- significant CpG sites or pathways were found, how- cular strength and is used to study age-related loss of ever two of the suggestive top CpG sites were mapped physical capacity. In order to explore the biological to the COL6A1 and CACNA1B genes, known to be mechanisms that influence hand grip strength varia- related to muscular dysfunction. By investigating tion, an epigenome-wide association study (EWAS) of genomic regions using the comb-p algorithm, several hand grip strength in 672 middle-aged and elderly differentially methylated regions in regulatory monozygotic twins (age 55–90 years) was performed, domains were identified as significantly associated to using both individual and twin pair level analyses, the hand grip strength, and pathway analyses of these latter controlling the influence of genetic variation. regions revealed significant pathways related to the Moreover, as measurements of hand grip strength immune system, autoimmune disorders, including performed over 8 years were available in the elderly diabetes type 1 and viral myocarditis, as well as twins (age 73–90 at intake), a longitudinal EWAS was negative regulation of cell differentiation. -
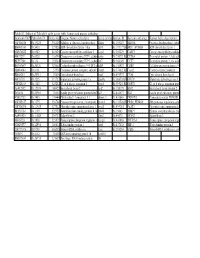
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

A Rare Variant in MCF2L Identified Using Exclusion Linkage in A
European Journal of Human Genetics (2016) 24, 86–91 & 2016 Macmillan Publishers Limited All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE A rare variant in MCF2L identified using exclusion linkage in a pedigree with premature atherosclerosis Stephanie Maiwald1,7, Mahdi M Motazacker1,7,8, Julian C van Capelleveen1, Suthesh Sivapalaratnam1, Allard C van der Wal2, Chris van der Loos2, John JP Kastelein1, Willem H Ouwehand3,4, G Kees Hovingh1, Mieke D Trip1,5, Jaap D van Buul6 and Geesje M Dallinga-Thie*,1 Cardiovascular disease (CVD) is a major cause of death in Western societies. CVD risk is largely genetically determined. The molecular pathology is, however, not elucidated in a large number of families suffering from CVD. We applied exclusion linkage analysis and next-generation sequencing to elucidate the molecular defect underlying premature CVD in a small pedigree, comprising two generations of which six members suffered from premature CVD. A total of three variants showed co-segregation with the disease status in the family. Two of these variants were excluded from further analysis based on the prevalence in replication cohorts, whereas a non-synonymous variant in MCF.2 Cell Line Derived Transforming Sequence-like protein (MCF2L, c.2066A4G; p.(Asp689Gly); NM_001112732.1), located in the DH domain, was only present in the studied family. MCF2L is a guanine exchange factor that potentially links pathways that signal through Rac1 and RhoA. Indeed, in HeLa cells, MCF2L689Gly failed to activate Rac1 as well as RhoA, resulting in impaired stress fiber formation. Moreover, MCF2L protein was found in human atherosclerotic lesions but not in healthy tissue segments. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

Novel Targets of Apparently Idiopathic Male Infertility
International Journal of Molecular Sciences Review Molecular Biology of Spermatogenesis: Novel Targets of Apparently Idiopathic Male Infertility Rossella Cannarella * , Rosita A. Condorelli , Laura M. Mongioì, Sandro La Vignera * and Aldo E. Calogero Department of Clinical and Experimental Medicine, University of Catania, 95123 Catania, Italy; [email protected] (R.A.C.); [email protected] (L.M.M.); [email protected] (A.E.C.) * Correspondence: [email protected] (R.C.); [email protected] (S.L.V.) Received: 8 February 2020; Accepted: 2 March 2020; Published: 3 March 2020 Abstract: Male infertility affects half of infertile couples and, currently, a relevant percentage of cases of male infertility is considered as idiopathic. Although the male contribution to human fertilization has traditionally been restricted to sperm DNA, current evidence suggest that a relevant number of sperm transcripts and proteins are involved in acrosome reactions, sperm-oocyte fusion and, once released into the oocyte, embryo growth and development. The aim of this review is to provide updated and comprehensive insight into the molecular biology of spermatogenesis, including evidence on spermatogenetic failure and underlining the role of the sperm-carried molecular factors involved in oocyte fertilization and embryo growth. This represents the first step in the identification of new possible diagnostic and, possibly, therapeutic markers in the field of apparently idiopathic male infertility. Keywords: spermatogenetic failure; embryo growth; male infertility; spermatogenesis; recurrent pregnancy loss; sperm proteome; DNA fragmentation; sperm transcriptome 1. Introduction Infertility is a widespread condition in industrialized countries, affecting up to 15% of couples of childbearing age [1]. It is defined as the inability to achieve conception after 1–2 years of unprotected sexual intercourse [2]. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein