A Rare Variant in MCF2L Identified Using Exclusion Linkage in A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In-Depth Analysis of Genetic Variation Associated with Severe West Nile Viral Disease
Article In-Depth Analysis of Genetic Variation Associated with Severe West Nile Viral Disease Megan E. Cahill 1, Mark Loeb 2, Andrew T. Dewan 1 and Ruth R. Montgomery 3,* 1 Center for Perinatal, Pediatric and Environmental Epidemiology, Department of Chronic Disease Epidemiology, Yale School of Public Health, 1 Church Street, New Haven, CT 06510, USA; [email protected] (M.E.C.); [email protected] (A.T.D.) 2 3208 Michael DeGroote Centre for Learning & Discovery, Division of Clinical Pathology, McMaster University, Hamilton, ON L8S 4L8, Canada; [email protected] 3 Department of Internal Medicine, Yale School of Medicine, 300 Cedar Street, New Haven, CT 06520, USA * Correspondence: [email protected] Received: 30 October 2020; Accepted: 3 December 2020; Published: 8 December 2020 Abstract: West Nile virus (WNV) is a mosquito-borne virus which causes symptomatic disease in a minority of infected humans. To identify novel genetic variants associated with severe disease, we utilized data from an existing case-control study of WNV and included population controls for an expanded analysis. We conducted imputation and gene-gene interaction analysis in the largest and most comprehensive genetic study conducted to date for West Nile neuroinvasive disease (WNND). Within the imputed West Nile virus dataset (severe cases n = 381 and asymptomatic/mild controls = 441), we found novel loci within the MCF.2 Cell Line Derived Transforming Sequence Like (MCF2L) gene (rs9549655 and rs2297192) through the individual loci analyses, although none reached statistical significance. Incorporating population controls from the Wisconsin Longitudinal Study on Aging (n = 9012) did not identify additional novel variants, a possible reflection of the cohort’s inclusion of individuals who could develop mild or severe WNV disease upon infection. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -
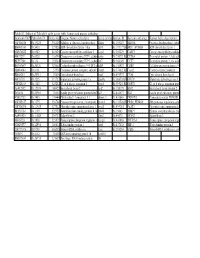
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

CD29 Identifies IFN-Γ–Producing Human CD8+ T Cells With
+ CD29 identifies IFN-γ–producing human CD8 T cells with an increased cytotoxic potential Benoît P. Nicoleta,b, Aurélie Guislaina,b, Floris P. J. van Alphenc, Raquel Gomez-Eerlandd, Ton N. M. Schumacherd, Maartje van den Biggelaarc,e, and Monika C. Wolkersa,b,1 aDepartment of Hematopoiesis, Sanquin Research, 1066 CX Amsterdam, The Netherlands; bLandsteiner Laboratory, Oncode Institute, Amsterdam University Medical Center, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; cDepartment of Research Facilities, Sanquin Research, 1066 CX Amsterdam, The Netherlands; dDivision of Molecular Oncology and Immunology, Oncode Institute, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands; and eDepartment of Molecular and Cellular Haemostasis, Sanquin Research, 1066 CX Amsterdam, The Netherlands Edited by Anjana Rao, La Jolla Institute for Allergy and Immunology, La Jolla, CA, and approved February 12, 2020 (received for review August 12, 2019) Cytotoxic CD8+ T cells can effectively kill target cells by producing therefore developed a protocol that allowed for efficient iso- cytokines, chemokines, and granzymes. Expression of these effector lation of RNA and protein from fluorescence-activated cell molecules is however highly divergent, and tools that identify and sorting (FACS)-sorted fixed T cells after intracellular cytokine + preselect CD8 T cells with a cytotoxic expression profile are lacking. staining. With this top-down approach, we performed an un- + Human CD8 T cells can be divided into IFN-γ– and IL-2–producing biased RNA-sequencing (RNA-seq) and mass spectrometry cells. Unbiased transcriptomics and proteomics analysis on cytokine- γ– – + + (MS) analyses on IFN- and IL-2 producing primary human producing fixed CD8 T cells revealed that IL-2 cells produce helper + + + CD8 Tcells. -

NBEAL2 Is Required for Neutrophil and NK Cell Function and Pathogen Defense
The Journal of Clinical Investigation BRIEF REPORT NBEAL2 is required for neutrophil and NK cell function and pathogen defense John M. Sowerby,1 David C. Thomas,1 Simon Clare,2 Marion Espéli,1,3 Jose A. Guerrero,4,5 Kim Hoenderdos,1 Katherine Harcourt,2 Morgan Marsden,6 Juneid Abdul-Karim,6 Mathew Clement,6 Robin Antrobus,7 Yagnesh Umrania,7 Philippa R. Barton,7 Shaun M. Flint,1 Jatinder K. Juss,1 Alison M. Condliffe,1 Paul A. Lyons,1 Ian R. Humphreys,6 Edwin R. Chilvers,1 Willem H. Ouwehand,2,4,5 Gordon Dougan,1,2 and Kenneth G.C. Smith1 1Department of Medicine, University of Cambridge School of Clinical Medicine, Cambridge Biomedical Campus, Cambridge, United Kingdom. 2Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, United Kingdom. 3INSERM UMR-996, Inflammation, Chemokines and Immunopathology, Université Paris-Sud, Université Paris-Saclay, Clamart, France. 4Department of Haematology, University of Cambridge, Cambridge Biomedical Campus, Cambridge, United Kingdom. 5NHS Blood and Transplant, Cambridge Biomedical Campus, Cambridge, United Kingdom. 6Division of Infection and Immunity, Cardiff University, Cardiff, United Kingdom. 7Cambridge Institute for Medical Research, Cambridge Biomedical Campus, Cambridge, United Kingdom. Mutations in the human NBEAL2 gene cause gray platelet syndrome (GPS), a bleeding diathesis characterized by a lack of α granules in platelets. The functions of the NBEAL2 protein have not been explored outside platelet biology, but there are reports of increased frequency of infection and abnormal neutrophil morphology in patients with GPS. We therefore investigated the role of NBEAL2 in immunity by analyzing the phenotype of Nbeal2-deficient mice. We found profound abnormalities in the Nbeal2-deficient immune system, particularly in the function of neutrophils and NK cells. -

NBEAL2 Gene Neurobeachin Like 2
NBEAL2 gene neurobeachin like 2 Normal Function The NBEAL2 gene provides instructions for making a protein whose function is unclear. The protein appears to be critical for the normal development of platelets, which are small blood cells involved in blood clotting. Platelets are produced in bone marrow, the spongy tissue in the center of long bones that produces most of the blood cells the body needs. Platelets are formed from large precursor cells known as megakaryocytes. Within these cells, the NBEAL2 protein is thought to play a role in the development of sacs called alpha-granules, which are the most abundant components of platelets. Alpha-granules contain growth factors and other proteins that are important for blood clotting and wound healing. In response to an injury that causes bleeding, the proteins stored in alpha-granules help platelets stick to one another to form a plug that seals off damaged blood vessels and prevents further blood loss. Health Conditions Related to Genetic Changes Gray platelet syndrome At least 35 mutations in the NBEAL2 gene have been found to cause gray platelet syndrome, a disorder associated with abnormal bleeding. Most people with gray platelet syndrome also develop a condition called myelofibrosis, which is characterized by the buildup of scar tissue (fibrosis) in the bone marrow that prevents it from making enough normal blood cells. Mutations in the NBEAL2 gene disrupt the normal production of alpha-granules in megakaryocytes. Without alpha-granules, platelets are abnormally large and fewer in number than usual (macrothrombocytopenia). The abnormal platelets also appear gray when viewed under a microscope, which gives this condition its name. -

Remodeling Adipose Tissue Through in Silico Modulation of Fat Storage For
Chénard et al. BMC Systems Biology (2017) 11:60 DOI 10.1186/s12918-017-0438-9 RESEARCHARTICLE Open Access Remodeling adipose tissue through in silico modulation of fat storage for the prevention of type 2 diabetes Thierry Chénard2, Frédéric Guénard3, Marie-Claude Vohl3,4, André Carpentier5, André Tchernof4,6 and Rafael J. Najmanovich1* Abstract Background: Type 2 diabetes is one of the leading non-infectious diseases worldwide and closely relates to excess adipose tissue accumulation as seen in obesity. Specifically, hypertrophic expansion of adipose tissues is related to increased cardiometabolic risk leading to type 2 diabetes. Studying mechanisms underlying adipocyte hypertrophy could lead to the identification of potential targets for the treatment of these conditions. Results: We present iTC1390adip, a highly curated metabolic network of the human adipocyte presenting various improvements over the previously published iAdipocytes1809. iTC1390adip contains 1390 genes, 4519 reactions and 3664 metabolites. We validated the network obtaining 92.6% accuracy by comparing experimental gene essentiality in various cell lines to our predictions of biomass production. Using flux balance analysis under various test conditions, we predict the effect of gene deletion on both lipid droplet and biomass production, resulting in the identification of 27 genes that could reduce adipocyte hypertrophy. We also used expression data from visceral and subcutaneous adipose tissues to compare the effect of single gene deletions between adipocytes from each -

Identification of Potential Pathogenic Genes Associated with Osteoporosis
610.BJBJR0010.1302/2046-3758.612.BJR-2017-0102 research-article2017 Freely available online OPEN ACCESS BJR RESEARCH Identification of potential pathogenic genes associated with osteoporosis Objectives B. Xia, Osteoporosis is a chronic disease. The aim of this study was to identify key genes in osteo- Y. Li, porosis. J. Zhou, Methods B. Tian, Microarray data sets GSE56815 and GSE56814, comprising 67 osteoporosis blood samples L. Feng and 62 control blood samples, were obtained from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were identified in osteoporosis using Limma pack- Jining No. 1 People’s age (3.2.1) and Meta-MA packages. Gene Ontology and Kyoto Encyclopedia of Genes and Hospital, Jining, Genomes enrichment analyses were performed to identify biological functions. Further- Shandong Province, more, the transcriptional regulatory network was established between the top 20 DEGs and China transcriptional factors using the UCSC ENCODE Genome Browser. Receiver operating char- acteristic (ROC) analysis was applied to investigate the diagnostic value of several DEGs. Results A total of 1320 DEGs were obtained, of which 855 were up-regulated and 465 were down- regulated. These differentially expressed genes were enriched in Gene Ontology terms and Kyoto Encyclopedia of Genes and Genomes pathways, mainly associated with gene expres- sion and osteoclast differentiation. In the transcriptional regulatory network, there were 6038 interactions pairs involving 88 transcriptional factors. In addition, the quantitative reverse transcriptase-polymerase chain reaction result validated the expression of several genes (VPS35, FCGR2A, TBCA, HIRA, TYROBP, and JUND). Finally, ROC analyses showed that VPS35, HIRA, PHF20 and NFKB2 had a significant diagnostic value for osteoporosis. -

Severe Hypertriglyceridemia in a Patient Heterozygous for a Lipoprotein Lipase Gene Allele with Two Novel Missense Variants
European Journal of Human Genetics (2015) 23, 1259–1261 & 2015 Macmillan Publishers Limited All rights reserved 1018-4813/15 www.nature.com/ejhg SHORT REPORT Severe hypertriglyceridemia in a patient heterozygous for a lipoprotein lipase gene allele with two novel missense variants Ursula Kassner*,1, Bastian Salewsky2,3, Marion Wühle-Demuth1, Istvan Andras Szijarto1, Thomas Grenkowitz1, Priska Binner4, Winfried März4,5,6, Elisabeth Steinhagen-Thiessen1,2 and Ilja Demuth*,2,3 Rare monogenic hyperchylomicronemia is caused by loss-of-function mutations in genes involved in the catabolism of triglyceride-rich lipoproteins, including the lipoprotein lipase gene, LPL. Clinical hallmarks of this condition are eruptive xanthomas, recurrent pancreatitis and abdominal pain. Patients with LPL deficiency and severe or recurrent pancreatitis are eligible for the first gene therapy treatment approved by the European Union. Therefore the precise molecular diagnosis of familial hyperchylomicronemia may affect treatment decisions. We present a 57-year-old male patient with excessive hypertriglyceridemia despite intensive lipid-lowering therapy. Abdominal sonography showed signs of chronic pancreatitis. Direct DNA sequencing and cloning revealed two novel missense variants, c.1302A4T and c.1306G4A, in exon 8 of the LPL gene coexisting on the same allele. The variants result in the amino-acid exchanges p.(Lys434Asn) and p.(Gly436Arg). They are located in the carboxy-terminal domain of lipoprotein lipase that interacts with the glycosylphosphatidylinositol- anchored HDL-binding protein (GPIHBP1) and are likely of functional relevance. No further relevant mutations were found by direct sequencing of the genes for APOA5, APOC2, LMF1 and GPIHBP1. We conclude that heterozygosity for damaging mutations of LPL may be sufficient to produce severe hypertriglyceridemia and that chylomicronemia may be transmitted in a dominant manner, at least in some families. -

Association of Genetic Variants with Atrial Fibrillation
178 BIOMEDICAL REPORTS 4: 178-182, 2016 Association of genetic variants with atrial fibrillation YUICHIRO YAMASE1, KIMIHIKO KATO2, HIDEKI HORIBE1, CHIKARA UEYAMA1, TETSUO FUJIMAKI3, MITSUTOSHI OGURI4, MASAZUMI ARAI5, SACHIRO WATANABE5, TOYOAKI MUROHARA6 and YOSHIJI YAMADA7 1Department of Cardiovascular Medicine, Gifu Prefectural Tajimi Hospital, Tajimi, Gifu 507‑8522; 2Department of Internal Medicine, Meitoh Hospital, Nagoya, Aichi 465‑0025; 3Department of Cardiovascular Medicine, Inabe General Hospital, Inabe, Mie 511‑0428; 4Department of Cardiology, Kasugai Municipal Hospital, Kasugai, Aichi 486‑8510; 5Department of Cardiology, Gifu Prefectural General Medical Center, Gifu, Gifu 500‑8717; 6Department of Cardiology, Nagoya University Graduate School of Medicine, Nagoya, Aichi 466‑8550; 7Department of Human Functional Genomics, Life Science Research Center, Mie University, Tsu, Mie 514‑8507, Japan Received October 1, 2015; Accepted November 27, 2015 DOI: 10.3892/br.2015.551 Abstract. Recent genome-wide association studies (GWASs) with atrial fibrillation, with the minor G and T alleles, respec- identified various genes and loci that confer susceptibility tively, representing risk factors for this condition. PSRC1 and to coronary artery disease or myocardial infarction among ZC3HC1 may thus be susceptibility loci for atrial fibrillation in Caucasian populations. As myocardial ischemia is an impor- Japanese individuals. tant risk factor for atrial fibrillation, we hypothesized that certain polymorphisms may contribute to the genetic suscep- -

Protein O-Glcnacylation Is Essential for the Maintenance of Renal Energy Homeostasis and Function Via Lipolysis During Fasting and Diabetes
BASIC RESEARCH www.jasn.org Protein O-GlcNAcylation Is Essential for the Maintenance of Renal Energy Homeostasis and Function via Lipolysis during Fasting and Diabetes Sho Sugahara,1 Shinji Kume,1 Masami Chin-Kanasaki,1,2 Issei Tomita,1 Mako Yasuda-Yamahara,1 Kosuke Yamahara,1 Naoko Takeda,1 Norihisa Osawa,1 Motoko Yanagita,3 Shin-ichi Araki,1,2 and Hiroshi Maegawa1 1Department of Medicine, Shiga University of Medical Science, Otsu, Shiga, Japan; 2Division of Blood Purification, Shiga University of Medical Science Hospital, Otsu, Shiga, Japan; and 3Department of Nephrology, Graduate School of Medicine, Kyoto University, Kyoto, Japan ABSTRACT Background Energy metabolism in proximal tubular epithelial cells (PTECs) is unique, because ATP pro- duction largely depends on lipolysis in both the fed and fasting states. Furthermore, disruption of renal lipolysis is involved in the pathogenesis of diabetic tubulopathy. Emerging evidence suggests that protein O-GlcNAcylation, an intracellular nutrient-sensing system, may regulate a number of metabolic pathways according to changes in nutritional status. Although O-GlcNAcylation in PTECs has been demonstrated experimentally, its precise role in lipolysis in PTECs is unclear. Methods To investigate the mechanism of renal lipolysis in PTECs—specifically, the role played by protein O-GlcNAcylation—we generated mice with PTECs deficient in O-GlcNAc transferase (Ogt). We analyzed their renal phenotypes during ad libitum feeding, after prolonged fasting, and after mice were fed a high- fat diet for 16 weeks to induce obesity and diabetes. Results Although PTEC-specific Ogt-deficient mice lacked a marked renal phenotype during ad libitum feeding, after fasting 48 hours, they developed Fanconi syndrome–like abnormalities, PTEC apoptosis, and lower rates of renal lipolysis and ATP production.