Single-Gene Deletions Contributing to Loss of Heterozygosity in Saccharomyces Cerevisiae: Genome-Wide Screens and Reproducibility
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

Molecular Basis for the Distinct Cellular Functions of the Lsm1-7 and Lsm2-8 Complexes
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.22.055376; this version posted April 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Molecular basis for the distinct cellular functions of the Lsm1-7 and Lsm2-8 complexes Eric J. Montemayor1,2, Johanna M. Virta1, Samuel M. Hayes1, Yuichiro Nomura1, David A. Brow2, Samuel E. Butcher1 1Department of Biochemistry, University of Wisconsin-Madison, Madison, WI, USA. 2Department of Biomolecular Chemistry, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA. Correspondence should be addressed to E.J.M. ([email protected]) and S.E.B. ([email protected]). Abstract Eukaryotes possess eight highly conserved Lsm (like Sm) proteins that assemble into circular, heteroheptameric complexes, bind RNA, and direct a diverse range of biological processes. Among the many essential functions of Lsm proteins, the cytoplasmic Lsm1-7 complex initiates mRNA decay, while the nuclear Lsm2-8 complex acts as a chaperone for U6 spliceosomal RNA. It has been unclear how these complexes perform their distinct functions while differing by only one out of seven subunits. Here, we elucidate the molecular basis for Lsm-RNA recognition and present four high-resolution structures of Lsm complexes bound to RNAs. The structures of Lsm2-8 bound to RNA identify the unique 2′,3′ cyclic phosphate end of U6 as a prime determinant of specificity. In contrast, the Lsm1-7 complex strongly discriminates against cyclic phosphates and tightly binds to oligouridylate tracts with terminal purines. -

Datasheet BA0931 Anti-HSPH1 Antibody
Product datasheet Anti-HSPH1 Antibody Catalog Number: BA0931 BOSTER BIOLOGICAL TECHNOLOGY Special NO.1, International Enterprise Center, 2nd Guanshan Road, Wuhan, China Web: www.boster.com.cn Phone: +86 27 67845390 Fax: +86 27 67845390 Email: [email protected] Basic Information Product Name Anti-HSPH1 Antibody Gene Name HSPH1 Source Rabbit IgG Species Reactivity human,rat,mouse Tested Application WB,IHC-P,ICC/IF,FCM Contents 500ug/ml antibody with PBS ,0.02% NaN3 , 1mg BSA and 50% glycerol. Immunogen A synthetic peptide corresponding to a sequence at the C-terminus of human Hsp105(713-733aa EVMEWMNNVMNAQAKKSLDQD), different from the related mouse sequence by one amino acid, and different from the related rat sequence by two amino acids. Purification Immunogen affinity purified. Observed MW 110KD Dilution Ratios Western blot: 1:500-2000 Immunohistochemistry in paraffin section IHC-(P): 1:50-400 Immunocytochemistry/Immunofluorescence (ICC/IF): 1:50-400 Flow cytometry (FCM): 1-3μg/1x106 cells (Boiling the paraffin sections in 10mM citrate buffer,pH6.0,or PH8.0 EDTA repair liquid for 20 mins is required for the staining of formalin/paraffin sections.) Optimal working dilutions must be determined by end user. Storage 12 months from date of receipt,-20℃ as supplied.6 months 2 to 8℃ after reconstitution. Avoid repeated freezing and thawing Background Information HSP105(HEAT-SHOCK 105/110-KD PROTEIN 1), also called HSPH1 or HSP110, is a protein that in humans is encoded by the HSPH1 gene. Immunohistochemical analysis localizes HSP105 mainly in the cytoplasm. Database analysis indicates that both HSP105 isoforms are highly conserved during evolution. -

Bppart and Bpmax: RNA-RNA Interaction Partition Function and Structure Prediction for the Base Pair Counting Model
BPPart and BPMax: RNA-RNA Interaction Partition Function and Structure Prediction for the Base Pair Counting Model Ali Ebrahimpour-Boroojeny, Sanjay Rajopadhye, and Hamidreza Chitsaz ∗ Department of Computer Science, Colorado State University Abstract A few elite classes of RNA-RNA interaction (RRI), with complex roles in cellular functions such as miRNA-target and lncRNAs in human health, have already been studied. Accordingly, RRI bioinfor- matics tools tailored for those elite classes have been proposed in the last decade. Interestingly, there are somewhat unnoticed mRNA-mRNA interactions in the literature with potentially drastic biological roles. Hence, there is a need for high-throughput generic RRI bioinformatics tools. We revisit our RRI partition function algorithm, piRNA, which happens to be the most comprehensive and computationally-intensive thermodynamic model for RRI. We propose simpler models that are shown to retain the vast majority of the thermodynamic information that piRNA captures. We simplify the energy model and instead consider only weighted base pair counting to obtain BPPart for Base-pair Partition function and BPMax for Base-pair Maximization which are 225 and 1350 faster ◦ × × than piRNA, with a correlation of 0.855 and 0.836 with piRNA at 37 C on 50,500 experimentally charac- terized RRIs. This correlation increases to 0.920 and 0.904, respectively, at 180◦C. − Finally, we apply our algorithm BPPart to discover two disease-related RNAs, SNORD3D and TRAF3, and hypothesize their potential roles in Parkinson's disease and Cerebral Autosomal Dominant Arteri- opathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL). 1 Introduction Since mid 1990s with the advent of RNA interference discovery, RNA-RNA interaction (RRI) has moved to the spotlight in modern, post-genome biology. -
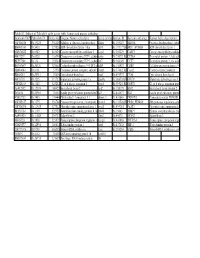
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

A Rare Variant in MCF2L Identified Using Exclusion Linkage in A
European Journal of Human Genetics (2016) 24, 86–91 & 2016 Macmillan Publishers Limited All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE A rare variant in MCF2L identified using exclusion linkage in a pedigree with premature atherosclerosis Stephanie Maiwald1,7, Mahdi M Motazacker1,7,8, Julian C van Capelleveen1, Suthesh Sivapalaratnam1, Allard C van der Wal2, Chris van der Loos2, John JP Kastelein1, Willem H Ouwehand3,4, G Kees Hovingh1, Mieke D Trip1,5, Jaap D van Buul6 and Geesje M Dallinga-Thie*,1 Cardiovascular disease (CVD) is a major cause of death in Western societies. CVD risk is largely genetically determined. The molecular pathology is, however, not elucidated in a large number of families suffering from CVD. We applied exclusion linkage analysis and next-generation sequencing to elucidate the molecular defect underlying premature CVD in a small pedigree, comprising two generations of which six members suffered from premature CVD. A total of three variants showed co-segregation with the disease status in the family. Two of these variants were excluded from further analysis based on the prevalence in replication cohorts, whereas a non-synonymous variant in MCF.2 Cell Line Derived Transforming Sequence-like protein (MCF2L, c.2066A4G; p.(Asp689Gly); NM_001112732.1), located in the DH domain, was only present in the studied family. MCF2L is a guanine exchange factor that potentially links pathways that signal through Rac1 and RhoA. Indeed, in HeLa cells, MCF2L689Gly failed to activate Rac1 as well as RhoA, resulting in impaired stress fiber formation. Moreover, MCF2L protein was found in human atherosclerotic lesions but not in healthy tissue segments. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Genomic Identification, Evolution and Sequence Analysis of the Heat
G C A T T A C G G C A T genes Article Genomic Identification, Evolution and Sequence Analysis of the Heat-Shock Protein Gene Family in Buffalo 1, 2, 3 4 1 Saif ur Rehman y, Asif Nadeem y , Maryam Javed , Faiz-ul Hassan , Xier Luo , Ruqayya Bint Khalid 3 and Qingyou Liu 1,* 1 State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, Guangxi University, Nanning 530005, China; [email protected] (S.u.R.); [email protected] (X.L.) 2 Department of Biotechnology, Virtual University of Pakistan, Lahore-54000, Pakistan; [email protected] 3 Institute of Biochemistry and Biotechnology, University of Veterinary and Animal Sciences, Lahore-54000, Pakistan; [email protected] (M.J.); [email protected] (R.B.K.) 4 Institute of Animal and Dairy Sciences, Faculty of Animal Husbandry, University of Agriculture, Faisalabad-38040, Pakistan; [email protected] * Correspondence: [email protected]; Tel.: +86-138-7880-5296 These authors contributed equally to this manuscript. y Received: 26 October 2020; Accepted: 18 November 2020; Published: 23 November 2020 Abstract: Heat-shock proteins (HSP) are conserved chaperones crucial for protein degradation, maturation, and refolding. These adenosine triphosphate dependent chaperones were classified based on their molecular mass that ranges between 10–100 kDA, including; HSP10, HSP40, HSP70, HSP90, HSPB1, HSPD, and HSPH1 family. HSPs are essential for cellular responses and imperative for protein homeostasis and survival under stress conditions. This study performed a computational analysis of the HSP protein family to better understand these proteins at the molecular level. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

FBXL19 Recruits CDK-Mediator to Cpg Islands of Developmental Genes Priming Them for Activation During Lineage Commitment
RESEARCH ARTICLE FBXL19 recruits CDK-Mediator to CpG islands of developmental genes priming them for activation during lineage commitment Emilia Dimitrova1, Takashi Kondo2†, Angelika Feldmann1†, Manabu Nakayama3, Yoko Koseki2, Rebecca Konietzny4‡, Benedikt M Kessler4, Haruhiko Koseki2,5, Robert J Klose1* 1Department of Biochemistry, University of Oxford, Oxford, United Kingdom; 2Laboratory for Developmental Genetics, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan; 3Department of Technology Development, Kazusa DNA Research Institute, Kisarazu, Japan; 4Nuffield Department of Medicine, TDI Mass Spectrometry Laboratory, Target Discovery Institute, University of Oxford, Oxford, United Kingdom; 5CREST, Japan Science and Technology Agency, Kawaguchi, Japan Abstract CpG islands are gene regulatory elements associated with the majority of mammalian promoters, yet how they regulate gene expression remains poorly understood. Here, we identify *For correspondence: FBXL19 as a CpG island-binding protein in mouse embryonic stem (ES) cells and show that it [email protected] associates with the CDK-Mediator complex. We discover that FBXL19 recruits CDK-Mediator to †These authors contributed CpG island-associated promoters of non-transcribed developmental genes to prime these genes equally to this work for activation during cell lineage commitment. We further show that recognition of CpG islands by Present address: ‡Agilent FBXL19 is essential for mouse development. Together this reveals a new CpG island-centric Technologies, Waldbronn, mechanism for CDK-Mediator recruitment to developmental gene promoters in ES cells and a Germany requirement for CDK-Mediator in priming these developmental genes for activation during cell lineage commitment. Competing interests: The DOI: https://doi.org/10.7554/eLife.37084.001 authors declare that no competing interests exist. -

Neuronal Vulnerability and Multilineage Diversity in Multiple Sclerosis
Europe PMC Funders Group Author Manuscript Nature. Author manuscript; available in PMC 2020 January 17. Published in final edited form as: Nature. 2019 June 13; 573(7772): 75–82. doi:10.1038/s41586-019-1404-z. Europe PMC Funders Author Manuscripts Neuronal vulnerability and multilineage diversity in multiple sclerosis Lucas Schirmer#1,2,3,4, Dmitry Velmeshev#1,5, Staffan Holmqvist2, Max Kaufmann6, Sebastian Werneburg7, Diane Jung1,5, Stephanie Vistnes1,4, John H. Stockley2, Adam Young8, Maike Steindel2,8, Brian Tung1,5,9, Nitasha Goyal1,5,9, Aparna Bhaduri1,5, Simone Mayer1,5, Jan Broder Engler6, Omer A. Bayraktar2, Robin J. M. Franklin8, Maximilian Haeussler10, Richard Reynolds11, Dorothy P. Schafer7, Manuel A. Friese6, Lawrence R. Shiow1,4, Arnold R. Kriegstein1,5,†, David H. Rowitch1,2,4,† 1Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research, University of California, San Francisco, San Francisco, CA 94143, USA 2Department of Paediatrics and Wellcome-MRC Cambridge Stem Cell Institute, University of Cambridge, Cambridge, CB2 0QQ, UK 3Department of Neurology, Medical Faculty Mannheim, University of Heidelberg, 68167 Mannheim, Germany 4Department of Pediatrics/Neonatology, University of California, San Francisco, San Francisco, CA 94158, USA 5Department of Neurology, University of California, San Francisco, San Francisco, CA 94158, Europe PMC Funders Author Manuscripts USA 6Institute of Neuroimmunology and Multiple Sclerosis, Center for Molecular Neurobiology Hamburg, University Medical Center Hamburg-Eppendorf,