Pharmacokinetic Characterization of Angiotensin Iv Analogs With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2014/0051633 A1 Harding Et Al
US 20140051633A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0051633 A1 Harding et al. (43) Pub. Date: Feb. 20, 2014 (54) HEPATOCYTE GROWTH FACTOR MIMICS (60) Provisional application No. 61/706,567, filed on Sep. AS THERAPEUTICAGENTS 27, 2012, provisional application No. 61/706,437, filed on Sep. 27, 2012. (71) Applicant: Washington State University, Pullman, WA (US) Publication Classification (72) Inventors: Joseph W. Harding, Pullman, WA (US); John W. Wright, Pullman, WA (US); (51) Int. C. Caroline C. Benoist, Nashville, TN C07K5/065 (2006.01) (US); Leen H. Kawas, Pullman, WA (52) U.S. C. (US); Gary A. Wayman, Pullman, WA CPC .................................. C07K5/06078 (2013.01) (US) USPC ........................................................... S14/95 (73) Assignee: Washington State University, Pullman, WA (US) (57) ABSTRACT (21) Appl. No.: 14/038,973 (22) Filed: Sep. 27, 2013 Small molecule, peptidic hepatocyte growth factors mimics, which act as both mimetics and antagonists, have been gen Related U.S. Application Data erated. These molecules have been shown or predicted to have (63) Continuation-in-part of application No. PCT/US2012/ therapeutic potential for numerous pathologies including 031815, filed on Apr. 2, 2012. dementia (e.g. Alzheimer's) and Parkinson's disease. Patent Application Publication Feb. 20, 2014 Sheet 1 of 40 US 2014/0051633 A1 a --- --- - - -s ------- CS -8- Scopolarine s -k- Oii exa-ow " -: N. Sks s i. Y s re. Dihexa-high as: ^ N.-- st-scs:- -------------------------------------------------------------------------------------------. Acquisition (days) Figure 1A -8-CSF -- -8- Scopoanine i. --- - -k- Dihexa-ow -- hexa-high reta-or 2 3 4 5 6 7 8 Acquisition (days) Figure 1B -8- vici-treated -- hexa Acquisition (days) Figure 1C Patent Application Publication Feb. -
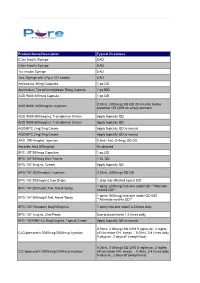
Pure Pricing List
Product Name/Description Typical Directions 0.3cc Insulin Syringe UAD 0.5cc Insulin Syringe UAD 1cc Insulin Syringe UAD 1mL Syringe with 27g x 1/2" needle UAD Amlexanox 40mg Capsules 1 po QD Ammonium Tetrathiomolybdate 50mg Capsule 1 po BID AOD 9604 600mcg Capsule 1 po QD 0.25mL (300mcg) SQ QD 30 minutes before AOD 9604 1200mcg/mL Injection breakfast OR QHS on empty stomach AOD 9604 600mcg/mL Transdermal Cream Apply topically QD AOD 9604 600mcg/mL Transdermal Cream Apply topically QD AOD/BPC 2mg/2mg Cream Apply topically QD to wound AOD/BPC 2mg/2mg Cream Apply topically QD to wound ARA 290 4mg/mL Injection 0.5mL-1mL (2-4mg) SQ QD Ascorbic Acid 500mg/mL As directed BPC-157 500mcg Capsules 1 po QD BPC-157 500mcg Mini-Troche 1 SL QD BPC-157 2mg/mL Cream Apply topically QD BPC-157 2000mcg/mL Injection 0.25mL (500mcg) SQ QD BPC-157 200mcg/mL Eye Drops 1 drop into affected eye(s) BID 1 spray (200mcg) into one nostril QD **Alternate BPC-157 200mcg/0.1mL Nasal Spray nostrils QD** 1 spray (500mcg) into one nostril QD-BID BPC-157 500mcg/0.1mL Nasal Spray **Alternate nostrils QD** BPC-157/ Synapsin 5mg/50mg/mL 1 spray into one nostril 2-3 times daily BPC-157 1mg/mL Oral Paste Use to brush teeth 1-2 times daily BPC-157/GHK-Cu 2mg/2mg/mL Topical Cream Apply topically QD to wound 0.05mL (100mcg) SQ QHS 5 nights on, 2 nights CJC/Ipamorelin 2000mcg/2000mcg Injection off (Increase GH, sleep)… 0.05mL 3-4 times daily 5 days on, 2 days off (weight loss) 0.05mL (100mcg) SQ QHS 5 nights on, 2 nights CJC/Ipamorelin 2000mcg/2000mcg Injection off (Increase GH, -

Counteracting EMF Toxicity with Peptides (Forget Global Warming Emfs Are Killing Us)
Counteracting EMF Toxicity with Peptides (Forget Global Warming EMFs are Killing Us) Dr. Kent Holtorf, MD Medical Director/CEO, Holtorf Medical Group (HoltorfMed.com) Medical Director/CEO, National Academy of Hypothyroidism (NAHypothyroidism.org) Medical Director, Integrative Peptides (IntegrativePeptides.com) Disclosure Statement Owner/CEO - Holtorf Medical Group HoltorfMed.com A multi-specialty medical group specializing in the treatment of complex endocrine dysfunction, CFS/fibromyalgia, chronic infectious diseases, immune dysfunction, neurodegenerative disease, and other complex chronic illnesses. Chief Medical Officer - The Non-Profit, The National Academy of Hypothyroidism and Integrative Sciences (NAHIS) NAHypothyroidism.org NAHIS is a non- profit, multidisciplinary medical society dedicated to the dissemination of new information on the diagnosis and treatment of hypothyroidism and complex conditions. NAHIS receives grants for clinical and laboratory research for novel methods in the diagnosis and treatment of hypothyroidism and chronic illnesses Chief Medical Officer - Integrative Peptides IntegrativePeptides.com Currently sells orally active and absorbable natural peptides in oral supplement form with unique delivery methods 2 Disclosure Statement Important Disclaimer In the interest of full disclosure, I am the All studies have limitations, and there is no perfect study. As with other studies, the studies Chief Medical Officer of Integrative Peptides. mentioned in this presentation and during the summit have limitations that should be considered when evaluating the efficacy of any treatment, including peptides, and determining the The opinions expressed are mine and do not appropriateness of peptide therapy for consumer use. A short summary, the abstract or reference to necessarily reflect those of Integrative a study may be discussed or provided. We are happy to provide you the entire study for your review of any study discussed, mentioned, presented or referenced. -

PEPTIDE INFORMATION Contact Kitt Burkley for Pricing 855-762-4878
PEPTIDE INFORMATION Contact Kitt Burkley for pricing 855-762-4878 AOD 9604 AOD 9604 works by mimicking the way natural HGH regulates fat metabolism but without the adverse effects on blood sugar or growth that is seen with unmodified HGH. Studies have suggested that AOD 9604 has an ability to stimulate lipolysis and inhibit lipogenesis. In addition AOD 9604 possesses other regenerative properties associated with growth hormone. Trials have been undertaken to determine the possible application of AOD 9604 in osteoarthritis and hypercholesterolemia, as well as for bone and cartilage repair. Results of oral glucose tolerance testing have demonstrated that AOD 9604 has no negative effect on carbohydrate metabolism, and it does not affect serum IGF-1 levels. AOD 9604 has an excellent safety profile and is generally well tolerated. It has attained Human GRAS status in the US. AOD 9604 600mcg/ml Cream – 30g AOD 9604* 0.5mg Troche BPC-157 (Body Protection Compound) BPC-157 promotes muscle and tendon healing, increases angiogenesis and decreases inflammatory response. Produces more type 1 collagen and has excellent potential for diabetic wound healing. BPC- 157 is highly stable, resistant to both hydrolysis and enzyme digestion. It can be given orally as well as administered subcutaneously or intramuscularly – no carrier molecule is required. This peptide has significant anti-inflammatory modulating effects. BPC-157 promotes tissue healing through signaling pathways – it provides specific response to particular injury thru cell signaling. BPC- 157 can be used continuously as body protective, or for specific period to treat body injury. BCP-157 is in clinical trials for IBD. -
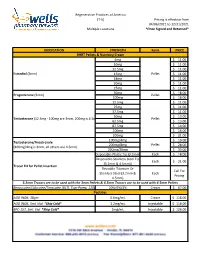
Formularies Pricing Sheet
Regenerative Practices of America (T-5) Pricing is effective from 04/06/2021 to 12/31/2021 Multiple Locations *Once Signed and Returned* MEDICATION STRENGTH Form PRICE BHRT Pellets & Numbing Cream 6mg $ 11.00 10mg $ 11.00 12.5mg $ 11.00 Estradiol (3mm) 15mg Pellet $ 11.00 18mg $ 11.00 20mg $ 11.00 25mg $ 11.00 50mg $ 18.00 Progesterone (3mm) Pellet 100mg $ 18.00 12.5mg $ 11.00 25mg $ 11.00 37.5mg $ 11.00 50mg $ 13.00 Testosterone (12.5mg - 100mg are 3mm, 200mg is 4.5mm) Pellet 62.5mg $ 13.00 87.5mg $ 14.00 100mg $ 16.00 200mg $ 21.00 100mg/4mg $ 19.00 Testosterone/Anastrozole 200mg/8mg Pellet $ 28.00 (100mg/4mg is 3mm, all others are 4.5mm) 200mg/20mg $ 33.00 Disposable-Plastic Tip (3.2mm) Each $ 18.00 Disposable-Stainless Steel Tip Each $ 21.00 (3.2mm & 4.5mm)) Trocar Kit for Pellet Insertion Reusable-Titanium Or Call For Stainless Steel (3.2mm & Each Pricing 4.5mm) 3.2mm Trocars are to be used with the 3mm Pellets & 4.5mm Trocars are to be used with 4.5mm Pellets Benzocaine/Lidocaine/Teracaine (BLT), Topi-Pump, 120gm 20%/6%/4% Cream $ 67.00 Peptides AOD 9604, 30gm 0.6mg/mL Cream $ 118.00 AOD 9604, 5mL Vial *Ship Cold* 1.2mg/mL Injectable $ 118.00 BPC-157, 5mL Vial *Ship Cold* 2mg/mL Injectable $ 126.00 Regenerative Practices of America (T-5) Pricing is effective from 04/06/2021 to 12/31/2021 Multiple Locations *Once Signed and Returned* 250mcg (0.25 mg) Capsule $ 74.00 BPC-157, Qty #30 *Ship Cold* 500mcg (0.5mg) Capsule $ 105.00 1mg Capsule $ 120.00 1mg/1mg/mL Injectable $ 55.00 CJC-1295/Ipamorelin, 2mL Vial *Ship Cold* 2mg/1mg/mL Injectable -

Product Data Sheet
Inhibitors Product Data Sheet Dihexa • Agonists Cat. No.: HY-16969 CAS No.: 1401708-83-5 Molecular Formula: C₂₇H₄₄N₄O₅ • Molecular Weight: 504.66 Screening Libraries Target: c-Met/HGFR Pathway: Protein Tyrosine Kinase/RTK Storage: Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month SOLVENT & SOLUBILITY In Vitro DMSO : 100 mg/mL (198.15 mM; Need ultrasonic) Mass Solvent 1 mg 5 mg 10 mg Concentration Preparing 1 mM 1.9815 mL 9.9077 mL 19.8153 mL Stock Solutions 5 mM 0.3963 mL 1.9815 mL 3.9631 mL 10 mM 0.1982 mL 0.9908 mL 1.9815 mL Please refer to the solubility information to select the appropriate solvent. In Vivo 1. Add each solvent one by one: 10% DMSO >> 40% PEG300 >> 5% Tween-80 >> 45% saline Solubility: ≥ 2.5 mg/mL (4.95 mM); Clear solution 2. Add each solvent one by one: 10% DMSO >> 90% corn oil Solubility: ≥ 2.5 mg/mL (4.95 mM); Clear solution BIOLOGICAL ACTIVITY Description Dihexa, an oligopeptide drug, is an orally active and blood-brain barrier-permeable angiotensin IV analog. Dihexa binds to hepatocyte growth factor (HGF) with high affinity (Kd=65 pM) and potentiates its activity at its receptor, c-Met. Dihexa exhibits excellent antidementia activity and improves cognitive function in animal models. Dihexa may have therapeutic potential as a treatment Alzheimer’s disease[1][2]. IC₅₀ & Target Kd: 65 pM (HGF)[1] In Vitro Dihexa binds with high affinity to HGF and both dihexa and its parent compound Norleucine 1-AngIV induce c-Met phosphorylation in the presence of subthreshold concentrations of HGF and augment HGF-dependent cell scattering. -

Product Catalogwells Pharmacy Network
2020 PRODUCT CATALOGWELLS PHARMACY NETWORK 800-622-4510 [email protected] wellsrx.com A COMPOUNDING PHARMACY DEDICATED CONTENT TO QUALITY, SERVICE AND QUESTIONS 1 SAFETY. 503B LICENSING 2 Wells Pharmacy Network is a privately held nationwide compounding pharmacy specializing in wellness, anti- aging, weight management, sexual wellness, aesthetic PRODUCTS dermatology and thyroid and adrenal health. LIST OF PRODUCTS 3 Our mission is to elevate compounding industry standards through a unique combination of superior science and service. This includes operating a 21,000 503A pharmacy that meet the patients specifi c compounding needs and as well as a 4,000 SF FDA Registered 503B Outsourcing Facility PELLETS that dispenses bulk medication for physician o ce use. ABOUT PELLETS 4 Wells Pharmacy Network uses the most advanced instrumentation, including in-house sterility and endotoxin STERILE 6 testing, which allow us to control the quality and timely IMPLANTABLE delivery of our preparations, such as sterile implantable HORMONE PELLETS bioidentical hormone pellets and other custom medications. Patient safety is our priority and our commitment to the quality of the preparations we compound is the most important thing we do each and every day. Learn more at: WELLSRX.COM CONTENT PEPTIDES AOD 9604 7 BPC-157 8 SEXUAL PRODUCTS CJC-1295/IPAMORELIN 9 WELLNESS DIHEXA 10 DSIP 11 PT-141 NASAL SPRAY 23 (EROS LOVE SPRAY) GHK-CU 12 SEXUAL WELLNESS 24 IBUTAMOREN 13 PROGRAM FOR IPAMORELIN 14 WOMEN KISSPEPTIN-10 15 SEXUAL WELLNESS 25 PROGRAM FOR MELATONAN II 16 -

Modified Polysaccharides, Method for Their Production and Their Use
(19) TZZ _¥_T (11) EP 2 899 213 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.07.2015 Bulletin 2015/31 C08B 1/00 (2006.01) C08B 1/06 (2006.01) C08B 11/00 (2006.01) C08B 15/06 (2006.01) (2006.01) (2006.01) (21) Application number: 14152669.9 C08B 31/00 C08B 31/08 C08B 37/00 (2006.01) C09D 101/08 (2006.01) (2006.01) (2006.01) (22) Date of filing: 27.01.2014 C09D 101/26 C09D 103/04 C09D 103/08 (2006.01) C09D 105/00 (2006.01) C09J 101/08 (2006.01) C09J 101/26 (2006.01) C09J 103/04 (2006.01) C09J 103/08 (2006.01) C09J 105/00 (2006.01) (84) Designated Contracting States: • Zajaczkowski-Fischer, Marta AL AT BE BG CH CY CZ DE DK EE ES FI FR GB 67141 Neuhofen (DE) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO • Kindler, Alois PL PT RO RS SE SI SK SM TR 67269 Grünstadt (DE) Designated Extension States: BA ME (74) Representative: Reitstötter Kinzebach Patentanwälte (71) Applicant: BASF SE Im Zollhof 1 67056 Ludwigshafen (DE) 67061 Ludwigshafen (DE) (72) Inventors: • Granström, Mari 00120 Helsinki (FI) (54) Modified polysaccharides, method for their production and their use (57) The present invention relates to a method for ride, ethylenically unsaturated polysaccharides obtaina- producing an ether modified polysaccharide, ether mod- ble by said method, polymers comprising those unsatu- ified polysaccharide obtainable by said method and the rated polysaccharides in polymerized form and the use use thereof. -

Tailor Made Compounding
PEPTIDE CATALOG TAILOR MADE COMPOUNDING Phone: 1 859 887 0013 | Email: [email protected] Our Story | 04-05 MK-677 | 36 Peptide Fact Sheets | 06-55 Myristyl | 37 3-Desoxy DHEA | 08 NMN | 38 5-Amino-1MQ | 09 PEG-MGF | 39 Amlexanox | 10 Pentosan Polysulfate | 40 AMT | 11 PNC-27 | 41 Aniracetam | 12 PT-141 | 42 AOD 9604 | 13 PTD-DBM | 43 AOD 9604 + HA | 14 RG3, Methylcobalamin, NAD+ | 44 BPC-157 | 15 Sarms LGD-4033 | 45 Cerebrolysin | 16 Selank | 46 CJC-1295 | 17 Semax | 47 DHH-B | 18 Tesamorelin | 48 Dihexa | 19 Tesofensine | 49 DSIP | 20 Tetradecylthioacetic Acid (TTA) | 50 Eclomiphene | 21 Thymosin Alpha-1 | 51 Epitalon | 22 Thymosin Beta | 52 FGL (l) | 23 VIP | 53 GHK-Cu | 24 Zinc Thymulin | 54 TABLE OF TABLE Glycyrrhetinic Acid & Aminophylline Dosing Charts | 25 56-62 Transdermal Fat Loss Cream Licensed States | 64-65 IGF-1 | 26 TMC App | 66-67 Ipamorelin | 27 Contact Us | 68 iRGD | 28 Kisspeptin-10 | 29 KPV | 30 Leuphasyl | 31 LL-37 | 32 Malanotan II | 33 Met-Enkephalin | 34 MOTS-c | 35 CONTENTS Tailor Made Compounding was launched United States. Since the inception of TMC, in the U.S. in January of 2016. We have we have sourced and compounded over 40 since become a major contributor to unique peptides. We are proud to be the peptide medicines in the integrative first in the United States to offer health space. compounds like CJC 1295, Bremelanotide, OUR PT 141, BPC-157, and Epitalon and Through our compounding expertise, continue to introduce new and exciting knowledge and experience, we have compounds to our formulary. -

AX Pharmaceutical Corp “We Supply High Quality Products All for Health”
AX Pharmaceutical Corp “We supply high quality products all for Health” Product List 1-Dodecanol 2,4-Dinitrochlorobenzene 2-Deoxy-D-Glucose 2-Methoxyestradiol 4-Aminopyridine 4-Methoxyphenol 5-Alpha-Hydroxy-Laxogenin 5-Amino-1-methylquinolinium 5-Aminolevulinic Acid Iodide 5-Aminosalicylic Acid 5-Fluororacil 5-Hydroxytryptophan Acebutolol HCl Acepromazine Maleate Acetazolamine Acetohydroxamic Acid Acetyl Dipeptide Acetyl Hexapeptide-3 Acetyl Tributyl Citrate ACTH 1-24 ACTH 1-39 Acyclovir Adapalene Adenosine 5 Monophosphate Adenosine 5 Tri-Phosphate Adenosylcobalamin Afoxolaner Disodium salt Alanyl-L-Glutamine Albendazole Alendronate Sodium Alendronic Acid Alpelisib Alpha Ketoglutaric Acid Alpha Lipoic Acid Alpha-Melanocyte-Stimulating Alprostadil Hormone Altrenogest Amantadine HCl Ambrisentan Ambroxol HCl Aminobenzoic Acid (PABA) Aminopentamide Sulfate Aminophylline Anhydrous Amitraz Amitriptyline HCl Amlexanox Amlodipine Besylate Amoxicillin Trihydrate Amphotericin B Anastrozole Aniracetam AOD 9604 Trifluoroacetate Aprepitant Aripiprazole Artemisinin Ascorbic Acid Ascorbyl Glucoside Atenolol Atipamezole HCl Atorvastatin Atovaquone Atropine Sulfate Monohydrate Auranofin Axitinib Azaperone Tartrate Azelaic Acid Azelastine HCl Azithromycin Dihydrate Azlocillin Sodium Bacitracin Zinc Baclofen Benazepril HCl Benzocaine Betadex (Beta Cyclodextrin) Betadex Sulfobutyl Ether Sodium Betahistine Bethanechol Chloride Bezafibrate Bifonazole Bimatoprost Bismuth Subsalicylate Bitter Orange Extract BPC 157 Bremelanotide Acetate (PT-141) Brimonidine -

Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes
cells Review Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes Sevda Gheibi *, Tania Singh, Joao Paulo M. C. M. da Cunha , Malin Fex and Hindrik Mulder * Unit of Molecular Metabolism, Lund University Diabetes Centre, Jan Waldenströms gata 35, Box 50332, SE-202 13 Malmö, Sweden; [email protected] (T.S.); [email protected] (J.P.M.C.M.d.C.); [email protected] (M.F.) * Correspondence: [email protected] (S.G.); [email protected] (H.M.) Received: 18 September 2020; Accepted: 9 November 2020; Published: 12 November 2020 Abstract: Type 2 diabetes, characterized by dysfunction of pancreatic β-cells and insulin resistance in peripheral organs, accounts for more than 90% of all diabetes. Despite current developments of new drugs and strategies to prevent/treat diabetes, there is no ideal therapy targeting all aspects of the disease. Restoration, however, of insulin-producing β-cells, as well as insulin-responsive cells, would be a logical strategy for the treatment of diabetes. In recent years, generation of transplantable cells derived from stem cells in vitro has emerged as an important research area. Pluripotent stem cells, either embryonic or induced, are alternative and feasible sources of insulin-secreting and glucose-responsive cells. This notwithstanding, consistent generation of robust glucose/insulin-responsive cells remains challenging. In this review, we describe basic concepts of the generation of induced pluripotent stem cells and subsequent differentiation of these into pancreatic β-like cells, myotubes, as well as adipocyte- and hepatocyte-like cells. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0068255 A1 Yu Et Al
US 20090068255A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0068255 A1 Yu et al. (43) Pub. Date: Mar. 12, 2009 (54) USE OF MATRIX METALLOPROTEINASE Publication Classification INHIBITORS IN SKIN CARE (51) Int. Cl. A6IR 8/14 (2006.01) (76) Inventors: Betty Yu, Cambridge, MA (US); A6IR 8/49 (2006.01) Amir Nashat, Newton, MA (US); A63L/35 (2006.01) Daniel Griffith Anderson, A61O 19/08 (2006.01) Sudbury, MA (US); David Thomas A61O 1704 (2006.01) Puerta, Melrose, MA (US); A6IP 7/02 (2006.01) Benjamin Adams, Cambridge, MA A6IP35/00 (2006.01) (US); Scott Clark, Pittsfield, MA A6IP 700 (2006.01) (US); Yushan Kim, Cambridge, A61O 19/00 (2006.01) MA (US); Eric George Spengler, A6II 3/442 (2006.01) Ridgefield, CT (US); Ronald P. A 6LX 9/27 (2006.01) McLaughlin, Reading, MA (US); (52) U.S. Cl. ............ 424/450; 514/459:514/350: 424/59 Susan Eilidh Bedford, Carlisle, (57) ABSTRACT MA (US); Zhi Li, San Diego, CA (US) The application of matrix metalloproteinase (MMP) inhibi tors to the skin inhibits the degradation of proteins found in the skin including collagen, elastin, and other basement mem Correspondence Address: brane and extracellular matrix protein. MMP inhibitors may FITZPATRICK CELLAHARPER & SCINTO be used in both cosmetic compositions and pharmaceutical 30 ROCKEFELLER PLAZA compositions for application to skin. MMP inhibitors are NEW YORK, NY 10112 (US) formulated with a cosmetically suitable vehicle or pharma ceutically acceptable excipient for application to the skin as (21) Appl. No.: 12/112,374 creams, lotions, ointments, Solutions, face masks, etc.