Insulin/Glucose-Responsive Cells Derived from Induced Pluripotent Stem Cells: Disease Modeling and Treatment of Diabetes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In Vitro Modulation of Cisplatin Accumulation in Human Ovarian Carcinoma Cells by Pharmacologic Alteration of Microtubules
In vitro modulation of cisplatin accumulation in human ovarian carcinoma cells by pharmacologic alteration of microtubules. R D Christen, … , D R Shalinsky, S B Howell J Clin Invest. 1993;92(1):431-440. https://doi.org/10.1172/JCI116585. Research Article We have previously shown that forskolin and 3-isobutyl-1-methylxanthine (IBMX) increased accumulation of cisplatin (DDP) in DDP-sensitive 2008 human ovarian carcinoma cells in proportion to their ability to increase cAMP. Since the major function of cAMP is to activate protein kinase A, it was conjectured that the stimulation of DDP accumulation was mediated by a protein kinase A substrate. We now show that exposure of 2008 cells to forskolin resulted in phosphorylation of a prominent 52-kD membrane protein. Microsequencing of the band demonstrated it to be human beta-tubulin. Similarly, pretreatment of 2008 cells with the microtubule stabilizing drug taxol increased platinum accumulation in a dose-dependent manner. In 11-fold DDP-resistant 2008/C13*5.25 cells, decreased DDP accumulation was associated with enhanced spontaneous formation of microtubule bundles and decreased expression of beta-tubulin and the tubulin-associated p53 antioncogene relative to 2008 cells. 2008/C13*5.25 cells had altered sensitivity to tubulin- binding drugs, being hypersensitive to taxol and cross-resistant to colchicine. We conclude that pharmacologic alterations of tubulin enhance accumulation of DDP, and that the DDP-resistant phenotype in 2008/C13*5.25 cells is associated with tubulin abnormalities. Find the latest version: https://jci.me/116585/pdf In Vitro Modulation of Cisplatin Accumulation in Human Ovarian Carcinoma Cells by Pharmacologic Alteration of Microtubules Randolph D. -

The Effects of Adenosine Antagonists on Distinct Aspects of Motivated Behavior: Interaction with Ethanol and Dopamine Depletion
Facultat de Ciències de la Salut Departament de Psicologia Bàsica, Clínica i Psicobiologia The effects of adenosine antagonists on distinct aspects of motivated behavior: interaction with ethanol and dopamine depletion. PhD Candidate Laura López Cruz Advisor Dr. Mercè Correa Sanz Co-advisor Dr. John D.Salamone Castelló, May 2016 Als meus pares i germà A Carlos AKNOWLEDGEMENTS This work was funded by two competitive grants awarded to Mercè Correa and John D. Salamone: Chapters 1-4: The experiments in the first chapters were supported by Plan Nacional de Drogas. Ministerio de Sanidad y Consumo. Spain. Project: “Impacto de la dosis de cafeína en las bebidas energéticas sobre las conductas implicadas en el abuso y la adicción al alcohol: interacción de los sistemas de neuromodulación adenosinérgicos y dopaminérgicos”. (2010I024). Chapters 5 and 6: The last 2 chapters contain experiments financed by Fundació Bancaixa-Universitat Jaume I. Spain. Project: “Efecto del ejercicio físico y el consumo de xantinas sobre la realización del esfuerzo en las conductas motivadas: Modulación del sistema mesolímbico dopaminérgico y su regulación por adenosina”. (P1.1B2010-43). Laura López Cruz was awarded a 4-year predoctoral scholarship “Fornación de Profesorado Universitario-FPU” (AP2010-3793) from the Spanish Ministry of Education, Culture and Sport. (2012/2016). TABLE OF CONTENTS ABSTRACT ...................................................................................................................1 RESUMEN .....................................................................................................................3 -

Phosphodiesterase Inhibitors Stimulate Osteoclast Formation Via TRANCE/RANKL Expression in Osteoblasts: Possible Involvement of ERK and P38 MAPK Pathways
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 579 (2005) 832–838 FEBS 29223 Phosphodiesterase inhibitors stimulate osteoclast formation via TRANCE/RANKL expression in osteoblasts: possible involvement of ERK and p38 MAPK pathways Masamichi Takamia,1, Eun Sook Chob,1, Soo Young Leec, Ryutaro Kamijoa, Mijung Yimb,* a Department of Biochemistry, School of Dentistry, Showa University, Tokyo 142-8555, Japan b College of Pharmacy, Sookmyung WomenÕs University, Seoul 140-742, Republic of Korea c Division of Molecular Life Sciences and Center for Cell Signaling Research, Ewha WomenÕs University, Seoul 120-750, Republic of Korea Received 20 October 2004; revised 1 December 2004; accepted 14 December 2004 Available online 12 January 2005 Edited by Lukas Huber several factors, such as 1,25-dihydroxyvitamin D Abstract Phosphodiesterases (PDEs) are enzymes that degrade 3 intracellular cAMP. In the present study, 3-isobutyl-1-methyl- [1,25(OH)2D3], parathyroid hormone (PTH), interleukin xanthine (IBMX) and pentoxifylline, PDE inhibitors, induced (IL)-6 plus soluble IL-6 receptor, prostaglandin E2 (PGE2), osteoclast formation in cocultures of mouse bone marrow cells and calcium [2–4]. Those factors induce the expression of and calvarial osteoblasts. These inhibitors induced the expression TNF-related activation-induced cytokine (TRANCE, also of the osteoclast differentiation factor, TNF-related activation known as RANKL, ODF, or OPGL) in osteoblasts, which induced cytokine (TRANCE, identical to RANKL, ODF, and triggers osteoclast differentiation [5–8]. Osteoblasts also pro- OPGL), in calvarial osteoblasts. IBMX induced phosphorylation duce osteoprotegerin (OPG), a decoy receptor for TRANCE, of extracellular signal-regulated kinase (ERK) and p38 mitogen- to inhibit osteoclast formation [9]. -

Exploring Cocoa Properties: Is Theobromine a Cognitive Modulator?
Psychopharmacology https://doi.org/10.1007/s00213-019-5172-0 REVIEW Exploring cocoa properties: is theobromine a cognitive modulator? Ilaria Cova1 & V. Leta1,2 & C. Mariani2 & L. Pantoni2 & S. Pomati1 Received: 7 May 2018 /Accepted: 16 January 2019 # Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Nutritional qualities of cocoa have been acknowledged by several authors; a particular focus has been placed on its high content of flavanols, known for their excellent antioxidant properties and subsequent protective effect on cardio- and cerebrovascular systems as well as for neuromodulatory and neuroprotective actions. Other active components of cocoa are methylxanthines (caffeine and theobromine). Whereas the effects of caffeine are extensively researched, the same is not the case for theobromine; this review summarizes evidence on the effect of theobromine on cognitive functions. Considering animal studies, it can be asserted that acute exposition to theobromine has a reduced and delayed nootropic effect with respect to caffeine, whereas both animal and human studies suggested a potential neuroprotective action of long-term assumption of theobromine through a reduction of Aβ amyloid pathology, which is commonly observed in Alzheimer’s disease patients’ brains. Hence, the conceiv- able action of theobromine alone and associated with caffeine or other cocoa constituents on cognitive modulation is yet underexplored and future studies are needed to shed light on this promising molecule. Keywords Cocoa . Theobromine . Cognitive modulator . Cognition Introduction predominant fraction of triglyceride molecules species (in particular oleic, stearic, palmitic, and linoleic acid) Theobroma cacao and its products (Pittenauer and Allmaier 2009); proteins contribute to 10–15% of the dry weight of cocoa seeds and consist Cocoa comes from the processing of seeds of a tropical tree, mainly of albumin and globulin fractions (Zak and considered by the Aztecs as a sacred plant. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0051633 A1 Harding Et Al
US 20140051633A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0051633 A1 Harding et al. (43) Pub. Date: Feb. 20, 2014 (54) HEPATOCYTE GROWTH FACTOR MIMICS (60) Provisional application No. 61/706,567, filed on Sep. AS THERAPEUTICAGENTS 27, 2012, provisional application No. 61/706,437, filed on Sep. 27, 2012. (71) Applicant: Washington State University, Pullman, WA (US) Publication Classification (72) Inventors: Joseph W. Harding, Pullman, WA (US); John W. Wright, Pullman, WA (US); (51) Int. C. Caroline C. Benoist, Nashville, TN C07K5/065 (2006.01) (US); Leen H. Kawas, Pullman, WA (52) U.S. C. (US); Gary A. Wayman, Pullman, WA CPC .................................. C07K5/06078 (2013.01) (US) USPC ........................................................... S14/95 (73) Assignee: Washington State University, Pullman, WA (US) (57) ABSTRACT (21) Appl. No.: 14/038,973 (22) Filed: Sep. 27, 2013 Small molecule, peptidic hepatocyte growth factors mimics, which act as both mimetics and antagonists, have been gen Related U.S. Application Data erated. These molecules have been shown or predicted to have (63) Continuation-in-part of application No. PCT/US2012/ therapeutic potential for numerous pathologies including 031815, filed on Apr. 2, 2012. dementia (e.g. Alzheimer's) and Parkinson's disease. Patent Application Publication Feb. 20, 2014 Sheet 1 of 40 US 2014/0051633 A1 a --- --- - - -s ------- CS -8- Scopolarine s -k- Oii exa-ow " -: N. Sks s i. Y s re. Dihexa-high as: ^ N.-- st-scs:- -------------------------------------------------------------------------------------------. Acquisition (days) Figure 1A -8-CSF -- -8- Scopoanine i. --- - -k- Dihexa-ow -- hexa-high reta-or 2 3 4 5 6 7 8 Acquisition (days) Figure 1B -8- vici-treated -- hexa Acquisition (days) Figure 1C Patent Application Publication Feb. -
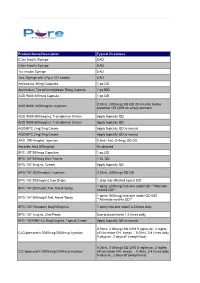
Pure Pricing List
Product Name/Description Typical Directions 0.3cc Insulin Syringe UAD 0.5cc Insulin Syringe UAD 1cc Insulin Syringe UAD 1mL Syringe with 27g x 1/2" needle UAD Amlexanox 40mg Capsules 1 po QD Ammonium Tetrathiomolybdate 50mg Capsule 1 po BID AOD 9604 600mcg Capsule 1 po QD 0.25mL (300mcg) SQ QD 30 minutes before AOD 9604 1200mcg/mL Injection breakfast OR QHS on empty stomach AOD 9604 600mcg/mL Transdermal Cream Apply topically QD AOD 9604 600mcg/mL Transdermal Cream Apply topically QD AOD/BPC 2mg/2mg Cream Apply topically QD to wound AOD/BPC 2mg/2mg Cream Apply topically QD to wound ARA 290 4mg/mL Injection 0.5mL-1mL (2-4mg) SQ QD Ascorbic Acid 500mg/mL As directed BPC-157 500mcg Capsules 1 po QD BPC-157 500mcg Mini-Troche 1 SL QD BPC-157 2mg/mL Cream Apply topically QD BPC-157 2000mcg/mL Injection 0.25mL (500mcg) SQ QD BPC-157 200mcg/mL Eye Drops 1 drop into affected eye(s) BID 1 spray (200mcg) into one nostril QD **Alternate BPC-157 200mcg/0.1mL Nasal Spray nostrils QD** 1 spray (500mcg) into one nostril QD-BID BPC-157 500mcg/0.1mL Nasal Spray **Alternate nostrils QD** BPC-157/ Synapsin 5mg/50mg/mL 1 spray into one nostril 2-3 times daily BPC-157 1mg/mL Oral Paste Use to brush teeth 1-2 times daily BPC-157/GHK-Cu 2mg/2mg/mL Topical Cream Apply topically QD to wound 0.05mL (100mcg) SQ QHS 5 nights on, 2 nights CJC/Ipamorelin 2000mcg/2000mcg Injection off (Increase GH, sleep)… 0.05mL 3-4 times daily 5 days on, 2 days off (weight loss) 0.05mL (100mcg) SQ QHS 5 nights on, 2 nights CJC/Ipamorelin 2000mcg/2000mcg Injection off (Increase GH, -

3-Isobutyl-1-Methylxanthine
Catalog Number: 195262 3-Isobutyl-1-methylxanthine Structure: Molecular Formula: C10H14N4O2 Formula Weight: 222.24 (as anhydrous) CAS #: 28822-58-4 Synonym: IBMX; MIX; MeiBu-Xan; IBX Solubility: Soluble in Krebs-Henseleit bicarbonate buffer, ethanol (10 mg/ml or 25 mg/ml with sonication17), DMSO (1 M with warming), or aqueous NaOH (pH 9.5); slightly soluble in water (0.3 mg/ml hot water). Solubility in 45% (w/v) aqueous 2-hydroxy-propyl--cyclodextrin is 3.2 mg/ml. Ethanol solutions can be stored at 2-8°C for approximately three (3) months.17 DMSO solutions should be aliquoted and stored at -20°C for 3 to 4 months. Aqueous solutions can be aliquoted and stored at -20°C for approximately 3 months.22 The aqueous solutions should be thawed for use by heating in a boiling water bath. Description: IBMX has been shown to be a potent, non-specific inhibitor of adenosine 3',5'-cyclic monophosphate phosphodiesterase (cAMP PDE)4, significantly more effective than theophylline.1,2,14,15,21 Also inhibits cGMP phosphodiesterases. IBMX inhibits cyclic nucleotide PDE with subsequent inhibition of cyclic nucleotide hydrolysis, resulting in accumulation of cyclic AMP and guanosine 3',5'-cyclic monophosphate.11,20 In a study of cyclic AMP and insulin release by islets of Langerhans, IBMX at 1 mM caused a marked increase in the intracellular concentration of cyclic AMP in the presence of glucose.14 IBMX, when used at 0.05 mM, was 20-fold more effective than theophylline at stimulating lipolysis in fat cells.2 It has been shown to promote the conversion of fibroblast cells into adipose cells, apparently without altering the amount of bromodeoxyuridine (BrdU) present in the DNA of the cells.16 The increase in cAMP level as a result of phosphodiesterase inhibition by IBMX activates PKA leading to decreased proliferation, increased differentiation, and induction of apoptosis.5,7,18 Other actions of IBMX: Inhibition of phenylephrine-induced release of 5-hydroxytryptamine from neuroendocrine epithelial cells of 9 the airway mucosa (IC50 = 1.3 uM). -

The Phosphodiesterase Inhibitor IBMX Blocks the Potassium Channel THIK-1 from the Extracellular Side
Molecular Pharmacology Fast Forward. Published on May 28, 2020 as DOI: 10.1124/molpharm.120.000011 This article has not been copyedited and formatted. The final version may differ from this version. The phosphodiesterase inhibitor IBMX blocks the potassium channel THIK-1 from the extracellular side Xinle Zou1, Linus J. Conrad1, Kristin Koschinsky, Günter Schlichthörl, Regina Preisig-Müller, Eugen Netz, Jens Krüger, Jürgen Daut2 and Downloaded from Vijay Renigunta2 1 Contributed equally molpharm.aspetjournals.org 2Corresponding authors Institute of Physiology and Pathophysiology, Marburg University, 35037 Marburg, Germany (X.Z., L.J.C., K.K, G.S., R.P.M., J.D., V.R.) at ASPET Journals on October 3, 2021 Biomolecular Interactions, Max Planck Institute for Developmental Biology, Tübingen, Germany (E.N.) High Performance and Cloud Computing Group, IT Center, University of Tübingen, Germany (J.K.) - 1 - Molecular Pharmacology Fast Forward. Published on May 28, 2020 as DOI: 10.1124/molpharm.120.000011 This article has not been copyedited and formatted. The final version may differ from this version. Running title: IBMX blocks THIK-1 Please send editorial correspondence to: Prof. Jürgen Daut Institute of Physiology and Pathophysiology Marburg University Deutschhausstr. 2 35037 Marburg, Germany Downloaded from Tel.: 0049-6421-2866494 Fax: 0049-6421-2866495 Email: [email protected]. molpharm.aspetjournals.org Text pages: 16 Tables: 0 Figures: 9 at ASPET Journals on October 3, 2021 Abstract: 215 words Introduction: 618 words Discussion: 1105 words Non-standard abbreviations CHO cells: Chinese hamster ovary cells IBMX: 3-isobutyl-1-methyl-xanthine THIK-1: Two-pore-domain halothane-inhibited K+ channel 1 THIK-2: Two-pore-domain halothane-inhibited K+ channel 2 TRAAK: TWIK-related arachidonic acid stimulated K+ channel TREK-1: TWIK-related K+ channel TWIK: tandem of P domains in a weakly inwardly rectifying K+ channel - 2 - Molecular Pharmacology Fast Forward. -

Counteracting EMF Toxicity with Peptides (Forget Global Warming Emfs Are Killing Us)
Counteracting EMF Toxicity with Peptides (Forget Global Warming EMFs are Killing Us) Dr. Kent Holtorf, MD Medical Director/CEO, Holtorf Medical Group (HoltorfMed.com) Medical Director/CEO, National Academy of Hypothyroidism (NAHypothyroidism.org) Medical Director, Integrative Peptides (IntegrativePeptides.com) Disclosure Statement Owner/CEO - Holtorf Medical Group HoltorfMed.com A multi-specialty medical group specializing in the treatment of complex endocrine dysfunction, CFS/fibromyalgia, chronic infectious diseases, immune dysfunction, neurodegenerative disease, and other complex chronic illnesses. Chief Medical Officer - The Non-Profit, The National Academy of Hypothyroidism and Integrative Sciences (NAHIS) NAHypothyroidism.org NAHIS is a non- profit, multidisciplinary medical society dedicated to the dissemination of new information on the diagnosis and treatment of hypothyroidism and complex conditions. NAHIS receives grants for clinical and laboratory research for novel methods in the diagnosis and treatment of hypothyroidism and chronic illnesses Chief Medical Officer - Integrative Peptides IntegrativePeptides.com Currently sells orally active and absorbable natural peptides in oral supplement form with unique delivery methods 2 Disclosure Statement Important Disclaimer In the interest of full disclosure, I am the All studies have limitations, and there is no perfect study. As with other studies, the studies Chief Medical Officer of Integrative Peptides. mentioned in this presentation and during the summit have limitations that should be considered when evaluating the efficacy of any treatment, including peptides, and determining the The opinions expressed are mine and do not appropriateness of peptide therapy for consumer use. A short summary, the abstract or reference to necessarily reflect those of Integrative a study may be discussed or provided. We are happy to provide you the entire study for your review of any study discussed, mentioned, presented or referenced. -

PEPTIDE INFORMATION Contact Kitt Burkley for Pricing 855-762-4878
PEPTIDE INFORMATION Contact Kitt Burkley for pricing 855-762-4878 AOD 9604 AOD 9604 works by mimicking the way natural HGH regulates fat metabolism but without the adverse effects on blood sugar or growth that is seen with unmodified HGH. Studies have suggested that AOD 9604 has an ability to stimulate lipolysis and inhibit lipogenesis. In addition AOD 9604 possesses other regenerative properties associated with growth hormone. Trials have been undertaken to determine the possible application of AOD 9604 in osteoarthritis and hypercholesterolemia, as well as for bone and cartilage repair. Results of oral glucose tolerance testing have demonstrated that AOD 9604 has no negative effect on carbohydrate metabolism, and it does not affect serum IGF-1 levels. AOD 9604 has an excellent safety profile and is generally well tolerated. It has attained Human GRAS status in the US. AOD 9604 600mcg/ml Cream – 30g AOD 9604* 0.5mg Troche BPC-157 (Body Protection Compound) BPC-157 promotes muscle and tendon healing, increases angiogenesis and decreases inflammatory response. Produces more type 1 collagen and has excellent potential for diabetic wound healing. BPC- 157 is highly stable, resistant to both hydrolysis and enzyme digestion. It can be given orally as well as administered subcutaneously or intramuscularly – no carrier molecule is required. This peptide has significant anti-inflammatory modulating effects. BPC-157 promotes tissue healing through signaling pathways – it provides specific response to particular injury thru cell signaling. BPC- 157 can be used continuously as body protective, or for specific period to treat body injury. BCP-157 is in clinical trials for IBD. -

Lack of Adipocyte Purinergic P2Y6 Receptor Greatly Improves Whole Body Glucose Homeostasis
Lack of adipocyte purinergic P2Y6 receptor greatly improves whole body glucose homeostasis Shanu Jaina, Sai P. Pydib, Kiran S. Totia, Bernard Robayec, Marco Idzkod,e, Oksana Gavrilovaf, Jürgen Wessb, and Kenneth A. Jacobsona,1 aMolecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD 20892; bMolecular Signaling Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD 20892; cInstitute of Interdisciplinary Research (IRIBHM), Université Libre de Bruxelles, 6041 Gosselies, Belgium; dUniversitätsklinik für Innere Medizin II, Klinische Abteilung für Pulmologie, Medizinische Universität, 1090 Vienna, Austria; eDepartment of Pneumology, University Hospital Freiburg, 79106 Freiburg, Germany; and fMouse Metabolism Core, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD 20892 Edited by Robert J. Lefkowitz, Howard Hughes Medical Institute, Durham, NC, and approved October 7, 2020 (received for review April 7, 2020) Uridine diphosphate (UDP)-activated purinergic receptor P2Y6 activation by uridine diphosphate (UDP) increases glucose uptake (P2Y6R) plays a crucial role in controlling energy balance through in adipocyte 3T3-L1 and skeletal muscle C2C12 cell lines (15). ’ central mechanisms. However, P2Y6R s roles in peripheral tissues P2Y6R also plays a pivotal role in inflammatory responses by regulating energy and glucose homeostasis remain unexplored. stimulating immune cell migration and inducing the secretion of Here, we report the surprising finding that adipocyte-specific de- inflammatory cytokines such as MCP1 and IL6 (16, 17). Stresses letion of P2Y6R protects mice from diet-induced obesity, improving such as environmental inflammation, chronic heart failure, and glucose tolerance and insulin sensitivity with reduced systemic in- dystrophic cardiomyopathy increase P2Y6R expression (18, 19). -

Camp Facilitates EDHF-Type Relaxations in Conduit Arteries by Enhancing Electrotonic Conduction Via Gap Junctions
cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions Tudor M. Griffith*, Andrew T. Chaytor, Hannah J. Taylor, Beverley D. Giddings, and David H. Edwards Department of Diagnostic Radiology, Wales Heart Research Institute, University of Wales College of Medicine, Heath Park, Cardiff CF14 4XN, United Kingdom Edited by Louis J. Ignarro, University of California, School of Medicine, Los Angeles, CA, and approved March 13, 2002 (received for review February 13, 2002) We have investigated the role of cAMP in NO- and prostanoid- endothelium-derived hyperpolarizing factor (EDHF) is released independent relaxations that are widely attributed to an endo- into the extracellular space by the endothelium on stimulation thelium-derived hyperpolarizing factor (EDHF). Under control con- with ACh to activate smooth muscle Kϩ channels and thereby ditions EDHF-type relaxations evoked by acetylcholine (ACh) in mediate relaxation (7, 8). However, bioassay experiments with rabbit iliac arteries were transient, but in the presence of the cAMP sandwich preparations of rabbit conduit arteries, constructed phosphodiesterase inhibitor isobutylmethylxanthine (IBMX) or the from closely apposed strips of endothelium-intact and -denuded cell permeant cAMP analog 8-bromo-cAMP, relaxations became vessel, fail to demonstrate the release of a freely diffusible sustained with their maxima potentiated Ϸ2-fold. Relaxation was EDHF after administration of ACh (9–11). Because there is no associated with transient Ϸ1.5-fold elevations in smooth muscle gap junctional communication between the ‘‘donor’’ endothe- cAMP levels with both mechanical and nucleotide responses being lium and ‘‘detector’’ smooth muscle in such vessel composites, abolished by interrupting gap junctional communication with the direct endothelial-smooth muscle coupling thus appears central connexin-mimetic peptide Gap 27 and by endothelial denudation.