Title: a TRANSMISSION-VIRULENCE EVOLUTIONARY TRADE-OFF
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catalogue of Clinical Trials and Cohort Studies to Identify Biological
Catalogue of clinical trials and cohort studies to identify biological specimens of relevance to the development of assays for acute and early HIV infection: Final Report Catalogue of clinical trials and cohort studies to identify biological specimens of relevance to the development of assays for recent HIV infection Final Report February 2010 This final report was prepared by Dr Joanne Micallef and Professor John Kaldor, The University of New South Wales, under subcontract with Family Health International, funded by the Bill and Melinda Gates Foundation under the grant titled “Development of Assays for Acute HIV Infection and Estimation and HIV Incidence in Population”. 1 Catalogue of clinical trials and cohort studies to identify biological specimens of relevance to the development of assays for acute and early HIV infection: Final Report Table of Contents Table of Contents .................................................................................................................................................................... 2 List of acronyms ...................................................................................................................................................................... 4 1 Introduction ..................................................................................................................................................................... 6 2 Methods ............................................................................................................................................................................ -

Download and Use
Shenkin et al. BMC Medicine (2019) 17:138 https://doi.org/10.1186/s12916-019-1367-9 RESEARCHARTICLE Open Access Delirium detection in older acute medical inpatients: a multicentre prospective comparative diagnostic test accuracy study of the 4AT and the confusion assessment method Susan D. Shenkin1, Christopher Fox2, Mary Godfrey3, Najma Siddiqi4, Steve Goodacre5, John Young6, Atul Anand7, Alasdair Gray8, Janet Hanley9, Allan MacRaild8, Jill Steven8, Polly L. Black8, Zoë Tieges1, Julia Boyd10, Jacqueline Stephen10, Christopher J. Weir10 and Alasdair M. J. MacLullich1* Abstract Background: Delirium affects > 15% of hospitalised patients but is grossly underdetected, contributing to poor care. The 4 ‘A’s Test (4AT, www.the4AT.com) is a short delirium assessment tool designed for routine use without special training. Theprimaryobjectivewastoassesstheaccuracyofthe4ATfor delirium detection. The secondary objective was to compare the 4AT with another commonly used delirium assessment tool, the Confusion Assessment Method (CAM). Methods: This was a prospective diagnostic test accuracy study set in emergency departments or acute medical wards involving acute medical patients aged ≥ 70. All those without acutely life-threatening illness or coma were eligible. Patients underwent (1) reference standard delirium assessment based on DSM-IV criteria and (2) were randomised to either the index test (4AT, scores 0–12; prespecified score of > 3 considered positive) or the comparator (CAM; scored positive or negative), in a random order, using computer-generated pseudo-random numbers, stratified by study site, with block allocation. Reference standard and 4AT or CAM assessments were performed by pairs of independent raters blinded to the results of the other assessment. Results: Eight hundred forty-three individuals were randomised: 21 withdrew, 3 lost contact, 32 indeterminate diagnosis, 2 missing outcome, and 785 were included in the analysis. -

Epidemiologic Study Designs
Epidemiologic Study Designs Jacky M Jennings, PhD, MPH Associate Professor Associate Director, General Pediatrics and Adolescent Medicine Director, Center for Child & Community Health Research (CCHR) Departments of Pediatrics & Epidemiology Johns Hopkins University Learning Objectives • Identify basic epidemiologic study designs and their frequent sequence of study • Recognize the basic components • Understand the advantages and disadvantages • Appropriately select a study design Research Question & Hypotheses Analytic Study Plan Design Basic Study Designs and their Hierarchy Clinical Observation Hypothesis Descriptive Study Case-Control Study Cohort Study Randomized Controlled Trial Systematic Review Causality Adapted from Gordis, 1996 MMWR Study Design in Epidemiology • Depends on: – The research question and hypotheses – Resources and time available for the study – Type of outcome of interest – Type of exposure of interest – Ethics Study Design in Epidemiology • Includes: – The research question and hypotheses – Measures and data quality – Time – Study population • Inclusion/exclusion criteria • Internal/external validity Epidemiologic Study Designs • Descriptive studies – Seeks to measure the frequency of disease and/or collect descriptive data on risk factors • Analytic studies – Tests a causal hypothesis about the etiology of disease • Experimental studies – Compares, for example, treatments EXPOSURE Cross-sectional OUTCOME EXPOSURE OUTCOME Case-Control EXPOSURE OUTCOME Cohort TIME Cross-sectional studies • Measure existing disease and current exposure levels at one point in time • Sample without knowledge of exposure or disease • Ex. Prevalence studies Cross-sectional studies • Advantages – Often early study design in a line of investigation – Good for hypothesis generation – Relatively easy, quick and inexpensive…depends on question – Examine multiple exposures or outcomes – Estimate prevalence of disease and exposures Cross-sectional studies • Disadvantages – Cannot infer causality – Prevalent vs. -

Systematic Review of the Prospective Cohort Studies on Meat Consumption and Colorectal Cancer Risk: a Meta-Analytical Approach1
Vol. 10, 439–446, May 2001 Cancer Epidemiology, Biomarkers & Prevention 439 Systematic Review of the Prospective Cohort Studies on Meat Consumption and Colorectal Cancer Risk: A Meta-Analytical Approach1 Manjinder S. Sandhu,2 Ian R. White, and confounded or modified by other factors cannot be Klim McPherson excluded. Department of Public Health and Primary Care, Institute of Public Health, University of Cambridge, Strangeways Research Laboratory, Cambridge, CB1 8RN [M. S. S.]; Medical Research Council Biostatistics Unit, Institute of Introduction Public Health, University of Cambridge, Cambridge, CB2 2SR [I. R. W.]; and The relation between meat consumption and colorectal cancer Cancer and Public Health Unit, London School of Hygiene and Tropical Medicine, London, WC1E 7HT [K. M.], United Kingdom risk remains controversial (1–10). Subsequent to the report of the National Academy of Sciences, “Diet and Health” (11), which implicated red meat as a causative factor in the etiology Abstract of colorectal cancer, two subsequent reports have reviewed the epidemiological evidence on meat and colorectal cancer risk (4, The relation between meat consumption and colorectal 3 cancer risk remains controversial. In this report, we 7). The report of the WCRF concluded: “The evidence shows quantitatively reviewed the prospective observational that red meat probably increases risk and processed meat pos- studies that have analyzed the relation between meat sibly increases risk of colorectal cancer” (7). The report from consumption and colorectal cancer. We conducted COMA judged that “there is moderately consistent evidence electronic searches of MEDLINE, EMBASE, and from cohort studies of a positive association between the con- CANCERLIT databases through to the end of June 1999 sumption of red or processed meat and risk of colorectal can- and manual searches of references from retrieved cer” (4). -

Overview of Epidemiological Study Designs
University of Massachusetts Medical School eScholarship@UMMS PEER Liberia Project UMass Medical School Collaborations in Liberia 2018-11 Overview of Epidemiological Study Designs Richard Ssekitoleko Yale University Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/liberia_peer Part of the Clinical Epidemiology Commons, Epidemiology Commons, Family Medicine Commons, Infectious Disease Commons, Medical Education Commons, and the Quantitative, Qualitative, Comparative, and Historical Methodologies Commons Repository Citation Ssekitoleko R. (2018). Overview of Epidemiological Study Designs. PEER Liberia Project. https://doi.org/ 10.13028/fp3z-mv12. Retrieved from https://escholarship.umassmed.edu/liberia_peer/5 This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in PEER Liberia Project by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. Overview of Epidemiological study Designs Richard Ssekitoleko Department of Global Health Yale University 1.1 Objectives • Understand the different epidemiological study types • Get to know what is involved in each type of study • Understand the strengths and Limitations of each study type Key terms • Population • Consists of all elements and is the group from which a sample is drawn • A sample is a subset of a population • Parameters • Summary data from a population • Statistics • Summary data from a sample • Validity • Extent to which a conclusion or statistic is well-founded and likely corresponds accurately to the parameter. Hierarchy of Evidence Study type Observational Interventional Descriptive Experiment Ecological Randomized Controlled Trial Cross-sectional Case-control Cohort Overview of Epidemiologic Study Designs Validity *anecdotes Cost Understanding What Physicians Mean: In my experience … once. -

Recommendations for Naming Study Design and Levels of Evidence
Recommendations for Naming Study Design and Levels of Evidence for Structured Abstracts in the Journal of Orthopedic & Sports Physical Therapy (JOSPT) Last revised 07-03-2008 The JOSPT as a journal that primarily focuses on clinically based research, attempts to assist readers in understanding the quality of research evidence in its published manuscripts by using a structured abstract that includes information on the “study design” and the classification of the “level of evidence.” While no single piece of information or classification can signify the quality and clinical relevance of published studies, a consistent approach to communicating this information is a step towards assisting our readers in evaluating new knowledge. Outline of the structured abstract for research reports published in JOSPT Study design Objectives Background Methods and Measures Results Conclusions Level of Evidence Suggestions for Naming Study Designs We recommend that authors attempt to use the following terminology when naming their research designs. Use of consistent terminology will make it easier for readers to identify the nature of the study and will allow us to track the type of studies published more easily. We recognize that this list is not all-inclusive and that more appropriate descriptors might be suitable for some studies. In those cases, investigators are encouraged to pick the most appropriate descriptors for their study. These suggestions are provided as a means of encouraging consistency where it would be both useful and informative and their use is expected where they do apply. Quantitative Clinical Study Categories include (Therapy, Prevention, Etiology, Harm, Prognosis, Diagnosis, Differential Diagnosis, Symptom Prevalence, Economic Analysis, and Decision Analysis). -

Lecture 13: Cohort Studies (Kanchanaraksa)
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike License. Your use of this material constitutes acceptance of that license and the conditions of use of materials on this site. Copyright 2008, The Johns Hopkins University and Sukon Kanchanaraksa. All rights reserved. Use of these materials permitted only in accordance with license rights granted. Materials provided “AS IS”; no representations or warranties provided. User assumes all responsibility for use, and all liability related thereto, and must independently review all materials for accuracy and efficacy. May contain materials owned by others. User is responsible for obtaining permissions for use from third parties as needed. Cohort Studies Sukon Kanchanaraksa, PhD Johns Hopkins University Design of a Cohort Study Identify: Exposed Not exposed follow: Develop Do not Develop Do not disease develop disease develop disease disease 3 Cohort Study Totals Exposed a + b First, identify Not c + d exposed 4 Cohort Study Then, follow to see whether Disease Disease does not develops develop Totals Exposed a b a + b First, identify Not c d c + d exposed 5 Cohort Study Calculate Then, follow to see whether and compare Disease Disease does not Incidence develops develop Totals of disease a Exposed a b a + b a+b First, identify Not c c d c + d exposed c+ d a c = Incidence in exposed = Incidence in not exposed a+ b c+ d 6 Cohort Study Then, follow to see whether Calculate Do not Develop develop Incidence CHD CHD Totals of disease Smoke 84 84 2916 3000 First, cigarettes 3000 select Do not 87 smoke 87 4913 5000 cigarettes 5000 84 = 0.028 = Incidence in 'smoke cigarettes' 3000 87 = 0.0174 = Incidence in 'not smoke cigarettes' 5000 7 Design of a Cohort Study Begin with: Defined population N o n - r a n d o m i z e d Identify : Exposed Not exposed follow: Develop Do not Develop Do not disease develop disease develop disease disease 8 Comparison of Experimental vs. -
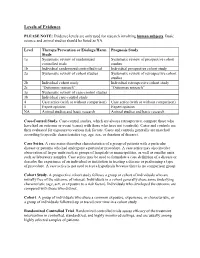
Levels of Evidence
Levels of Evidence PLEASE NOTE: Evidence levels are only used for research involving human subjects. Basic science and animal studies should be listed as NA. Level Therapy/Prevention or Etiology/Harm Prognosis Study Study 1a Systematic review of randomized Systematic review of prospective cohort controlled trials studies 1b Individual randomized controlled trial Individual prospective cohort study 2a Systematic review of cohort studies Systematic review of retrospective cohort studies 2b Individual cohort study Individual retrospective cohort study 2c “Outcomes research” “Outcomes research” 3a Systematic review of case-control studies 3b Individual case-control study 4 Case series (with or without comparison) Case series (with or without comparison) 5 Expert opinion Expert opinion NA Animal studies and basic research Animal studies and basic research Case-Control Study. Case-control studies, which are always retrospective, compare those who have had an outcome or event (cases) with those who have not (controls). Cases and controls are then evaluated for exposure to various risk factors. Cases and controls generally are matched according to specific characteristics (eg, age, sex, or duration of disease). Case Series. A case series describes characteristics of a group of patients with a particular disease or patients who had undergone a particular procedure. A case series may also involve observation of larger units such as groups of hospitals or municipalities, as well as smaller units such as laboratory samples. Case series may be used to formulate a case definition of a disease or describe the experience of an individual or institution in treating a disease or performing a type of procedure. A case series is not used to test a hypothesis because there is no comparison group. -

Saliva Exhibits High Sensitivity and Specificity for the Detection
diseases Review Saliva Exhibits High Sensitivity and Specificity for the Detection of SARS-COV-2 Ibrahim Warsi 1,2 , Zohaib Khurshid 3,* , Hamda Shazam 4, Muhammad Farooq Umer 5 , Eisha Imran 6 , Muhammad Owais Khan 7, Paul Desmond Slowey 8,9 and J. Max Goodson 2,10 1 Medical Sciences in Clinical Investigation, Harvard Medical School, Boston, MA 02115, USA; [email protected] 2 The Forsyth Institute, Cambridge, MA 02142, USA; [email protected] 3 Department of Prosthodontics and Dental Implantology, College of Dentistry, King Faisal University, Al-Ahsa 31982, Saudi Arabia 4 Department of Oral Pathology, College of Dentistry, Ziauddin University, Karachi 74700, Pakistan; [email protected] 5 Al-Shifa School of Public Health, Al-Shifa Trust, Jhelum Road, Rawalpindi, Punjab 46000, Pakistan; [email protected] 6 Department of Dental Materials, Institute of Medical Sciences, HITEC Dental College, Taxilla 751010, Pakistan; [email protected] 7 Dow International Dental College (DIDC), Dow University of Health Sciences, Karachi 74200, Pakistan; [email protected] 8 Health Science Center, School of Public Health, Xi’an Jiaotong University, Xi’an 710061, China; [email protected] 9 Oasis Diagnostics®Corporation, Vancouver, WA 98686, USA 10 Department of Oral Medicine, Infection, and Immunology, Harvard School of Dental Medicine, Boston, MA 02115, USA Citation: Warsi, I.; Khurshid, Z.; * Correspondence: [email protected]; Tel.: +966-558-420-410 Shazam, H.; Umer, M.F.; Imran, E.; Khan, M.O.; Slowey, P.D.; Goodson, Abstract: In the wake of the COVID-19 pandemic, it is crucial to assess the application of a multitude J.M. Saliva Exhibits High Sensitivity of effective diagnostic specimens for conducting mass testing, for accurate diagnosis and to formulate and Specificity for the Detection of strategies for its prevention and control. -

Cohort, Cross Sectional, and Case-Control Studies C J Mann
54 RESEARCH SERIES Emerg Med J: first published as 10.1136/emj.20.1.54 on 1 January 2003. Downloaded from Observational research methods. Research design II: cohort, cross sectional, and case-control studies C J Mann ............................................................................................................................. Emerg Med J 2003;20:54–60 Cohort, cross sectional, and case-control studies are While an appropriate choice of study design is collectively referred to as observational studies. Often vital, it is not sufficient. The hallmark of good research is the rigor with which it is conducted. A these studies are the only practicable method of checklist of the key points in any study irrespec- studying various problems, for example, studies of tive of the basic design is given in box 1. aetiology, instances where a randomised controlled trial Every published study should contain suffi- cient information to allow the reader to analyse might be unethical, or if the condition to be studied is the data with reference to these key points. rare. Cohort studies are used to study incidence, causes, In this article each of the three important and prognosis. Because they measure events in observational research methods will be discussed with emphasis on their strengths and weak- chronological order they can be used to distinguish nesses. In so doing it should become apparent between cause and effect. Cross sectional studies are why a given study used a particular research used to determine prevalence. They are relatively quick method and which method might best answer a particular clinical problem. and easy but do not permit distinction between cause and effect. Case controlled studies compare groups COHORT STUDIES retrospectively. -

Asymptomatic SARS-Cov-2 Infection: a Systematic Review and Meta-Analysis
Asymptomatic SARS-CoV-2 infection: A systematic review and meta-analysis Pratha Saha, Meagan C. Fitzpatricka,b, Charlotte F. Zimmera, Elaheh Abdollahic, Lyndon Juden-Kellyc, Seyed M. Moghadasc, Burton H. Singerd,1, and Alison P. Galvania aCenter for Infectious Disease Modeling and Analysis, Yale School of Public Health, New Haven, CT 06520; bCenter for Vaccine Development and Global Health, University of Maryland School of Medicine, Baltimore, MD 21201; cAgent-Based Modelling Laboratory, York University, Toronto, ON M3J 1P3, Canada; and dEmerging Pathogens Institute, University of Florida, Gainesville, FL 32610 Contributed by Burton H. Singer, July 8, 2021 (sent for review May 19, 2021; reviewed by David Fisman and Claudio Jose Struchiner) Quantification of asymptomatic infections is fundamental for 20%. Two methodological issues limit the accuracy of these studies. effective public health responses to the COVID-19 pandemic. Dis- First, pooled asymptomaticity reported in these studies is likely bi- crepancies regarding the extent of asymptomaticity have arisen ased downward because they did not account for study designs from inconsistent terminology as well as conflation of index and which have a higher representation of cases experiencing symptoms secondary cases which biases toward lower asymptomaticity. We (10). Second, one of the meta-analyses (7) did not consider biases in searched PubMed, Embase, Web of Science, and World Health Or- reported asymptomaticity that can arise from inadequate longitu- ganization Global Research Database on COVID-19 between Janu- dinal follow-up. Studies that assess the symptom profile only at the ary 1, 2020 and April 2, 2021 to identify studies that reported silent time of testing or do not follow up symptoms for a sufficiently long infections at the time of testing, whether presymptomatic or time period cannot distinguish presymptomatic from asymptomatic asymptomatic. -

Bias in Research
A Review Paper Bias in Research Steven S. Agabegi, MD, and Peter J. Stern, MD types of bias in such studies include selection bias, perfor- Abstract mance bias, attrition bias and detection bias.5,6 Other terms Bias is a systematic inconsistency in research that con- have been used to describe the types of bias (eg, informa- taminates the primary comparison. There are several tion bias, recall bias, ascertainment bias), but the above 4 forms of bias, and there are specific methods of mini- categories are the most common and will be discussed. In mizing them in different study designs. The randomized this article, we will describe the forms of bias and how they controlled trial (RCT) is the gold standard to which all can be minimized in different study designs. other study designs are compared. However, errors can be made at various stages of a RCT that introduce bias. Furthermore, not all questions can be addressed by a TYPES OF STUDIES RCT, and in some cases another study design may be The advantages and disadvantages of 4 types of study more appropriate. Observational studies are more prone designs are summarized in Table I. to bias, but, when properly conducted with rigorous Case reports and case series provide anecdotal infor- methods to minimize bias, these studies can be valuable mation for rare conditions or new treatments, determine in clinical research. long-term outcomes of a procedure, and describe the natu- ral history of a condition or the complication rates after ias is a systematic inconsistency in research stud- a surgical procedure.