Introduction of Fluorine and Fluorine- Containing Functional Groups
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
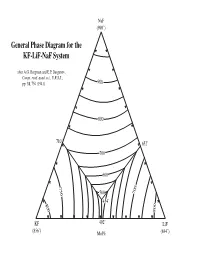
General Phase Diagram for the KF-Lif-Naf System
NaF (990˚) General Phase Diagram for the KF-LiF-NaF System after A.G. Bergman and E.P. Dergunov, Compt. rend. acad. sci., U.R.S.S., pp. 31, 754 (1941). 900 800 710˚ 652˚ 700 600 750 500 700 454˚ 800 800 KF 492˚ LiF (856˚) Mol% (844˚) Sodium Fluoride NaF 950˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 940˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 930˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 920˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 910˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 900˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 890˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 880˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 870˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 860˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 850˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 840˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 830˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 820˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 810˚ 810˚ ˚ KF 810 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 800˚ 800˚ ˚ KF 800 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 790˚ 790˚ ˚ KF 790 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 780˚ 780˚ ˚ LiF KF 780 Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 770˚ 770˚ ˚ LiF KF 770 Potassium Fluoride -

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -
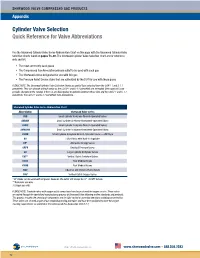
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

United States Patent Office Patented July 1, 1969
3,453,337 United States Patent Office Patented July 1, 1969 1. 2 3,453,337 FLUORINATION OF HALOGENATED The presence in the reaction mixture of the two fluo ORGANIC COMPOUNDS rides, or the complex fluoride enables better yields of Royston Henry Bennett and David Walter Cottrell, Ayon highly fluorinated products to be obtained under less mouth, England, assignors to Imperial Smelting Cor severe reaction condions, markedly increases the amount poration (N.S.C.) Limited, London, England, a British of fluorination reagent reacted under otherwise similar company conditions and enables the fluorination reaction to be No brawing. Filed Feb. 19, 1965, Ser. No. 434,128 carried out (for the same yield of product) at a lower Claims priority, application Great Britain, Feb. 26, 1964, temperature with the consequent use of less costly ma 7,932/64 terials and techniques of reactor construction. The pres Int, C. C07c 25/04 10 ence of the fluorides enables the vapor phase reaction U.S. C. 260-650 2 Claims to be carried out (for the same yields) at lower pressure This invention relates to the fluorination of organic than the pressure involved in the reactions using only halogen compounds and more especially to a process the alkali metal fluorides as proposed hitherto. The fur for the production of highly fluorinated aromatic com ther possibility of using a continuous flow apparatus such pounds by the replacement of higher halogen atoms in 15 as a fluidised reactor will be apparent to those familiar halogeno-aromatic compounds by fluorine atoms. with the art. The presence of the two fluorides or com Aromatic halogenocarbons containing carbon and halo plex fluoride enables a lower temperature to be em gen atoms only can be reacted with alkali fluorides ployed than was hitherto believed to be necessary, with under various conditions to give yields of halofluoro a consequent reduction in the extent of thermal degrada aromatic compounds. -
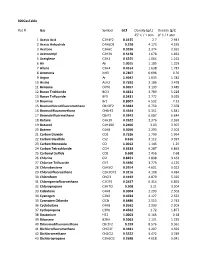
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Reaction of Potassium Fluoride with Organic Halogen Compounds. I
Reaction of Potassium Fluoride with Organic Halogen Compounds. I) Reactions of Potassium Fluoride with Organic Halides, Acids, aad Esters in presence ef Dimethyl Formamide and their Pyrolytic Decaboxylation in presence of Potassium Fluoride By You Sun Kim Atomic Energy Research Institute, Korea 有機 할로겐 化合物과 弗化加里의 反應 (第1報) 有機 할라어드, 酸 및 에스테르와 弗化加里의 디메칠 호쁨아마이드 溶蝶系 反應 및 高混■■脫炭酸-熱分解反應 金 裕 *善 (1963. 6. 19 受理) Abstract Reactions between potassium fluride with organic halogen-containning carboxylic acids in dimethyl formamide solvent gave a decarboxylation reaction for the case of fluoro carboxylic acids of the type of CF3 COOH, C3F7COOH, and C2F5COOH, whereas an additional partial fluorination together with dimeri zation reaction occured for the chlorine containning acids of the type of CH2CICOOH, CH3CHCICOOH, CHCI2COOH and o-Cl-CeHi-COOH. The phenyl halides showed no reactivity, but the halides with two electron attracting substituents on the benzene ring gave mainly dimerization reaction. The esters and alcohols gave an usual fluorination reaction. The same reactions in absence of the solvent at the elevated temperature increase the yield of the dimerized product and gave the cyclized product, fluorenone, in case of o-chlorobenzoic acid. It was found that the fluorination usually precede the decarboxylation reaction by checking the stiochemical sequence of reaction. Catalytic influence of potassium fluoride were discussed and the mechanism of the reaction was considered. 耍 約 「디메望호름아마이드」溶媒系에서 有機含할로겐化合物을 弗化加里와 反應시켜 본 結果 CFsCOOH, CsF’COOH, CzFQOOH 와 같은 含弗素有機酸에서는 脫炭酸反應이 일어나며, 含鹽素有機酸, CH2C1COOH. CH3CHC1COOH, CHC12- COOH 및 o-CK사LCOOH 은 一部 弗化反應이 일어 나고 雙合어imerization) 反應이 隨伴된다는 것을 究明하였다. -

Fluorinated Butatrienes
Fluorinated Butatrienes Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) eingereicht im Fachbereich Biologie, Chemie, Pharmazie der Freien Universität Berlin vorgelegt von Dipl.-Chem. Christian Ehm aus Berlin Berlin, April 2010 1. Gutachter: Prof. Dr. Dieter Lentz 2. Gutachter: Prof. Dr. Beate Paulus Disputation am 28.6.2010 I Acknowledgements It would not have been possible to write this doctoral thesis without the help and support of the kind people around me, to only some of whom it is possible to give particular mention here. First and foremost I would like to thank my principal supervisor, Professor Dieter Lentz, for the opportunity of doing research in his group. Without his continuous support and encouragement this thesis would not be in the present state. I highly appreciate that Professor Beate Paulus has agreed to be co-referee of my thesis. I would like to cordially thank Lada for her love and patience as well as her interest in my research. Special thanks to my family for their continuous support and love. I would like to thank Mike Roland, Sten Dathe and Sven Wünsche for their friendship and the fun we have had every Sunday evening. Special thanks to Sebastian Freitag, Boris Bolsinger and Frederic Heinrich for their friendship. They deserve much gratefulness for keeping me on the right way. I would like to thank all my colleagues at the Institut für Chemie und Biochemie, Abteilung Anorganische Chemie. In particular I want to thank all members of the Lentz group, Thomas Hügle, Moritz Kühnel, Dr. Floris Akkerman, Dr. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Hazardous Materials Descriptions and Codes
2012 Commodity Flow Survey Hazardous Materials Descriptions and Codes Hazardous Materials Descriptions and Proper Shipping Names UN or NA Code Accellerene, see p-Nitrosodimethylaniline Accumulators, electric, see Batteries, wet etc Accumulators, pressurized, pneumatic or hydraulic (containing non-flammable gas), see Articles pressurized, pneumatic or hydraulic (containing non-flammable gas) Acetal 1088 Acetaldehyde 1089 Acetaldehyde ammonia 1841 Acetaldehyde oxime 2332 Acetic acid, glacial or Acetic acid solution, with more than 80 percent acid, by mass 2789 Acetic acid solution, not less than 50 percent but not more than 80 percent acid, by 2790 mass Acetic acid solution, with more than 10 percent and less than 50 percent acid, by mass 2790 Acetic anhydride 1715 Acetone 1090 Acetone cyanohydrin, stabilized 1541 Acetone oils 1091 Acetonitrile 1648 Acetyl bromide 1716 Acetyl chloride 1717 Acetyl iodide 1898 Acetyl methyl carbinol 2621 Acetylene, dissolved 1001 Acetylene tetrabromide, see Tetrabromoethane Acid butyl phosphate, see Butyl acid phosphate Acid, sludge, see Sludge acid Acridine 2713 Acrolein dimer, stabilized 2607 Acrolein, stabilized 1092 Acrylamide, solid 2074 Acrylamide solution 3426 Acrylic acid, stabilized 2218 Acrylonitrile, stabilized 1093 Actuating cartridge, explosive, see Cartridges, power device Adhesives, containing a flammable liquid 1133 Adiponitrile 2205 Aerosols, poison, Packing Group III (each not exceeding 1 L capacity) 1950 Aerosols, flammable, (each not exceeding 1 L capacity) 1950 Source: Electronic Code of Federal Regulations http://ecfr.gpoaccess.gov/cgi/t/text/text- idx?c=ecfr&sid=dfec99df9f21ef29b59a8565433a56cd&rgn=div6&view=text&node=49:2.1.1.3.8.2&idno=49 1 Hazardous Materials Descriptions and Proper Shipping Names UN or NA Code Aerosols, flammable, n.o.s. -

Effects of Potassium and Sodium Fluoride in Different
Dental Materials Journal 2020; : – Effects of potassium and sodium fluoride in different concentrations on micro- shear bond strength and inhibition of demineralization Ali AL-QAHTANI1, Go INOUE1, Ahmed ABDOU1, Toru NIKAIDO2 and Junji TAGAMI1 1 Department of Cariology and Operative Dentistry, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), 1-5-45 Yushima, Bunkyo-ku, Tokyo, Japan 2 Department of Operative Dentistry, Division of Oral Functional Science and Rehabilitation, School of Dentistry, Asahi University, Hozumi 1851, Mizuho, Gifu 501-0296, Japan Corresponding author, Go INOUE; E-mail: [email protected] The aim of this study was to investigate the effects of potassium fluoride (KF) and sodium fluoride (NaF) in different concentrations on micro-shear bond strength (µSBS) and their protective effects against acid. The enamel blocks were treated with several concentrations of KF and NaF. For µSBS, Clearfil SE Bond 2 was applied to the treated surface and resin composite was light-cured, then examined using a universal testing machine. For acid resistance test, the specimens were immersed in acidic solution (pH 4.5), then examined under 3D confocal laser scanning microscope (CLSM). In µSBS, KF at 1,000, 9,000, and 10,000 ppm did not show differences compared with the control, while other concentrations of KF and NaF led to decreased µSBS. Higher concentrations of NaF and KF showed higher resistance to the acid challenge. So, we concluded that various concentrations of KF and NaF solutions had specific effects on µSBS and acid resistance. Keywords: Potassium fluoride, Sodium fluoride, Micro shear bonding strength, Acid resistance protective effect against erosive enamel loss compared INTRODUCTION with tooth mousses casein phosphopeptide-amorphous Dental caries is a degradation of tooth structure, which calciumphosphate (CPP-ACP)11). -

United States Patent Office E 8
United States Patent Office e 8. P t t 8 d s ep t. 6, 9 C 2 The addition compounds of phosphorus oxychloride 2,951,742 with niobium pentachloride and/or tantalum pentachlo PROCESS FOR THE RECOVERY OF METAL ride so obtained are solid compounds at ordinary tem HALDES FROM THER ADDUCTSWTH perature, which have lower melting points than those PHOSPHORUs oxYCHLORIDE of the pentachlorides used as starting materials. The constitution of the addition products has not been fully Waitersigao Scheier,to Ciba Neuewel, Limited, nearBasel, Basel, Switzerlaid, Switzerland, a Swiss as ascertained. However, analysis, has shown that there fina are formed, inter alia, 1:1-adducts of the metal penta chlorides with phosphorus oxychloride. The adducts are No Drawing. Filed Nov. 8, 1957, Ser. No. 695,229 O generally prepared industrially from mixtures of chlori Ciains priority, application Switzerland Nov. 13, 1956 nation products which are obtained, for example, by the chlorination of materials which contain niobium and 18 Claims. (C. 23-87) tantalum in oxidised form, for example, slags or espe cially concentrates or- ores, which may be after-treated This invention provides a process for the recovery 5 for the purpose of enrichment, or by the chlorination of of metal halides, especially chlorides of metals of the a mixtures of oxides of the aforesaid metals. The chlori fifth group of the periodic system, by splitting adducts nation of the aforesaid materials is carried out with of the metal halides with phosphorus oxychloride. chlorine gas and a reducing agent, such as carbon, or In patent application No. -

Potassium Fluoride As a Base in Organic Reactions
POTASSIUM FLUORIDE AS A BASE IN ORGANIC REACTIONS SOLUBILIZED BY 18-CROWN-6 A THESIS Presented to The Faculty of the Division of Graduate Studies by Thomas Ray Henson In Partial Fulfillment of the Requirements for the Degree Master of Science in Chemistry Georgia Institute of Technology March, 1975 POTASSIUM FLUORIDE AS A BASE IN ORGANIC REACTIONS SOLUBILIZED BY 18-CROWN-6 Approved: Charles Liotta, Chairman in^/ Grovenstein Date approved by Chairman £ UJgyuly 11*75 ACKNOWLEDGMENTS The author wishes to express his appreciation to his research director, Dr. C. L. Liotta, for the suggestion of this research problem and for his guidance and encouragement throughout the course of this work. The author also wishes to express his appreciation to Drs. E. Grovenstein and L. Zalkow for reading this thesis. The Department of Chemistry is gratefully acknowledged for financial support, as well as. Dr. C. L. Liotta. Finally, the author expresses a special word of thanks to his wife, Sandy, whose understanding and encouragement made this work possible. iii TABLE OF CONTENTS Page ACKNOWLEDGMENTS ii LIST OF TABLES iv LIST OF ILLUSTRATIONS v SUMMARY vi Chapter I. INTRODUCTION 1 Condensation Reactions Potassium Fluoride as a Base Crown Ethers II. EXPERIMENTAL 13 Chemicals 18-Crown-6 Synthesis Michael Condensations Alkylations 18-Crown-6 Complexes III. RESULTS AND DISCUSSIONS 47 Michael Condensations Knoevenagel Condensations Alkylations 18-Crown-6 Complexes IV. CONCLUSIONS 66 V. RECOMMENDATIONS 67 BIBLIOGRAPHY 68 iv LIST OF TABLES Table Page 1. Solubility of KF by 18-Crown-6 at 25° C 48 2. Michael Condensations Initiated by Potassium Fluoride in the Presence and Absence of 18-Crown-6 49 3.