Fluorinated Butatrienes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deoxygenation Supplemental Without Spectra
Novel Organic Transformations Arising from Gold(I) Chemistry By Mathieu André Morin Thesis submitted to the faculty of Graduate and Postdoctoral Studies In partial fulfillment of the requirements for the Ph.D. degree in chemistry Candidate Supervisor Mathieu André Morin Dr. Louis Barriault Ottawa-Carleton Chemistry Institute Faculty of Science University of Ottawa © Mathieu André Morin, Ottawa, Canada, 2017 Abstract The use of gold in organic chemistry is a relatively recent occurrence. In addition to being previously considered too expensive, it was also believed to be chemically inert. Soon after the early reports indicating its rich reactivity, the number of reports on chemical transformations involving gold sky rocketed. One such report by Toste and coworkers demonstrated the intramolecular addition of silyl enol ether onto Au(I) activated alkynes, resulting in a 5-exo dig cyclization. The first part (Chapter 1) of this thesis discusses the development of a Au(I) catalyzed polycyclization reaction inspired by this transformation. The reaction demonstrated the ability of Au(I) to successfully catalyze the formation of multiple C-C bonds and resulted in the synthesis of benzothiophenes, benzofurans, carbazoles and hydrindene. With the current resurgence of photochemical transformations being reported in literature, various opportunities for the use of Au(I) complexes arose. The substantial relativistic effect observed in gold which make it a good soft Lewis acid also has a significant influence on the redox potential of this metal. Chapter 3 of this thesis discusses the development of a Au(I) photocatalyzed process which benefits from having both a strong oxidation and reduction potential for the reduction of carbon-halide bonds. -

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

New Applications of Cyclobutadiene Cycloadditions: Diversity and Target Oriented Synthesis
New Applications of Cyclobutadiene Cycloadditions: Diversity and Target Oriented Synthesis Author: Jason Joseph Marineau Persistent link: http://hdl.handle.net/2345/1741 This work is posted on eScholarship@BC, Boston College University Libraries. Boston College Electronic Thesis or Dissertation, 2010 Copyright is held by the author, with all rights reserved, unless otherwise noted. Boston College The Graduate School of Arts and Sciences Department of Chemistry NEW APPLICATIONS OF CYCLOBUTADIENE CYCLOADDITIONS: DIVERSITY AND TARGET ORIENTED SYNTHESIS a dissertation by JASON JOSEPH MARINEAU submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy December 2010 © Copyright by JASON JOSEPH MARINEAU 2010 New Applications of Cyclobutadiene Cycloadditions: Diversity and Target Oriented Synthesis Jason J. Marineau Thesis Advisor: Professor Marc. L. Snapper Abstract Cyclobutadiene cycloadditions provide rapid access to rigid polycyclic systems with high strain energy and unusual molecular geometries. Further functionalization of these systems allows entry into unexplored chemical space. A tricarbonylcyclobutadiene iron complex on solid support enables exploration of these cycloadditions in a parallel format amenable to diversity oriented synthesis. Modeling of the cycloaddition transition states with density functional calculations provides a theoretical basis for analysis of the regioselectivity observed in generation of these substituted bicyclo[2.2.0]hexene derivatives. The high strain energy accessible in cyclobutadiene cycloadducts and their derivatives renders them useful synthons for access to medium-ring natural products through ring expansion. Torilin, a guaiane sesquiterpene isolated from extracts of the fruits of Torilis japonica, exhibits a range of biological activities including testosterone 5 -reductase inhibition, hKv1.5 channel blocking, hepatoprotective, anti-inflammatory and anti-cancer effects. -

CHEMISTRY 412/512 MIDTERM # 1 – Answer Key February 14, 2007 1
CHEMISTRY 412/512 MIDTERM # 1 – answer key February 14, 2007 1. (8 pts) Acyl cations ( RCO ) are key intermediates in Friedel – Crafts acylation reactions. They are stabilized by the presence of oxygen and this can be demonstrated through either resonance or MO analysis. a. Consider the formyl cation, shown below. Show how it is stabilized, using appropriate resonance structures. HCO formyl cation HCO HCO Acyl cations are stabilized by conjugation of one of the lone pairs at the oxygen center. b. Construct the orbital mixing diagram for the formyl cation, starting with group orbitals for CH and O-atom. Consider only first order mixing. Place the appropriate number of valence electrons and demonstrate how QMOT explains the stabilization of the cation. O HC The mixing diagram clearly shows the formation of TWO equivalent π-bonds, which is analogous to the resonance structure on the right, i.e. there is significant contribution by the O-atom. 2. (6 pts) Antiaromatic molecules avoid antiaromaticity by structural distortion. For example, cyclobutadiene is not square (and antiaromatic) but rectangular. Show, using π-group orbital analysis, that such distortion would indeed be stabilizing. square rectangular Solution: Instability of square cyclobutadiene is due to the presence of two degenerate, half-filled π-molecular orbitals (π2 and π3). In the rectangular form, qualitatively, the MOs remain the same, but the strength of some bonding/antibonding interactions is affected and it causes the degeneracy to be lifted. Focus on π2 and π3. Upon stretching, the antibonding interaction in π2 is reduced, while the same geometric distortion leads to reduction of the bonding interaction in π3. -
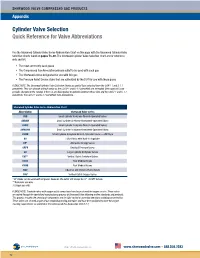
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

An Indicator of Triplet State Baird-Aromaticity
inorganics Article The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity Rabia Ayub 1,2, Kjell Jorner 1,2 ID and Henrik Ottosson 1,2,* 1 Department of Chemistry—BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden; [email protected] (R.A.); [email protected] (K.J.) 2 Department of Chemistry-Ångström Laboratory Uppsala University, Box 523, SE-751 20 Uppsala, Sweden * Correspondence: [email protected]; Tel.: +46-18-4717476 Received: 23 October 2017; Accepted: 11 December 2017; Published: 15 December 2017 Abstract: Baird’s rule tells that the electron counts for aromaticity and antiaromaticity in the first ππ* triplet and singlet excited states (T1 and S1) are opposite to those in the ground state (S0). Our hypothesis is that a silacyclobutene (SCB) ring fused with a [4n]annulene will remain closed in the T1 state so as to retain T1 aromaticity of the annulene while it will ring-open when fused to a [4n + 2]annulene in order to alleviate T1 antiaromaticity. This feature should allow the SCB ring to function as an indicator for triplet state aromaticity. Quantum chemical calculations of energy and (anti)aromaticity changes along the reaction paths in the T1 state support our hypothesis. The SCB ring should indicate T1 aromaticity of [4n]annulenes by being photoinert except when fused to cyclobutadiene, where it ring-opens due to ring-strain relief. Keywords: Baird’s rule; computational chemistry; excited state aromaticity; Photostability 1. Introduction Baird showed in 1972 that the rules for aromaticity and antiaromaticity of annulenes are reversed in the lowest ππ* triplet state (T1) when compared to Hückel’s rule for the electronic ground state (S0)[1–3]. -

Synthesis of Alkylated Benzenes and Lithium Aluminium Hydride Reduction
70 - 14,004 DAUERNHEIM, Lauren William, 1938- SYNTHESIS OF ALKYLATED BENZENES AND LITHIUM ALUMINUM HYDRIDE REDUCTION OF 1,3-DIHALIDES. The Ohio State University, Ph.D. , 1969 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan THIS DISSERTATION HAS BEEN MICROFILMED EXACTLY AS RECEIVED SYNTHESIS OP ALKYLATED BENZENES AND LITHIUM ALUMINUM HYDRIDE REDUCTION OF 1,3-DIHALIDES DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Lauren William Dauernheim, B.A., M.A ****** The Ohio State University 1969 Approved by Adviser Department of Chemistry ACKNOWLEDGMENT I am thankful for having had the opportunity to work under Dr. Melvin S. Newman. His guidance and support made fulfillment of this research possible. ii VITA February 16, 1938 , ....... Born, St. Louis, Missouri 1960 ...................... A.B., Washington University St. Louis, Missouri 1963 M.A., University of Arizona Tucson, Arizona iii TABLE OF CONTENTS Page ACKNOWLEDGMENTS ................................... ii VITA ............................................. iii LIST OF TABLES.................................... v LIST OF FIGURES ................................... vi LIST OF C H A R T S ......... vii PART I. SYNTHETIC ROUTES TO CERTAIN ALKYLATED BENZENES INTRODUCTION ................................ 1 SYNTHETIC ROUTES TO ALKYLATED NEOPENTYLBENZENES . 12 SYNTHETIC ROUTES TO ALKYLATED ETHYLBENZENES ........ 20 PART II. SYNTHESIS AND LITHIUM ALUMINUM HYDRIDE REDUCTION OF CERTAIN 1,3-DIHALOPROPANES INTRODUCTION ..................................... 23 SYNTHESIS OF 2-BENZYL-2-METHYL-1,3-DIHALOPROPANES . 25 SYNTHESIS OF 2-BENZYL-l,3-DIHALOPROPANES .......... 27 DISCUSSION OF THE NMR SPECTRA OF 2-BENZYL-l,3- DIHALOPROPANES ................................. 29 RESULTS AND CONCLUSIONS FROM THE LITHIUM ALUMINUM HYDRIDE REDUCTION OF CERTAIN 1,3-DIHALOPROPANES . 33 EXPERIMENTAL OF PART I .............. -

A Biomimetic Approach to Okilactomycin and Chrolactomycin and the Hexadehydro-Diels–Alder (HDDA) Reaction
A Biomimetic Approach to Okilactomycin and Chrolactomycin and The Hexadehydro-Diels–Alder (HDDA) Reaction A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY Dawen Niu IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Thomas R. Hoye December 2013 © Dawen Niu 2013 Acknowledgements So many things could have happened differently in the past 27 years and if any of them had, my career would have taken totally different pathways. I am so glad to be where I am today and need to acknowledge many people that made me here. First of all, I thank my parents. Born in remote villages, neither of them was able to receive good school education. They have been making very little money for what they do. To pay my (and my sister’s) tuition, they chose to work extremely hard. While I was in college, my father worked ca. 12 hours a day and >300 days a year even though he was suffering severe back pain. They tried all they could to make ends meet and yet never complained. But they started to complain after I left for US and stopped asking for tuition. They complain because they aren’t sure if I could get used to the American lifestyle and because they fear my feet will be dragged in US. “Mother worries when son travels a thousand miles.” This recent five years passed really fast to me, but it must have been the longest five years to them. I understand all these better after being a parent myself. -
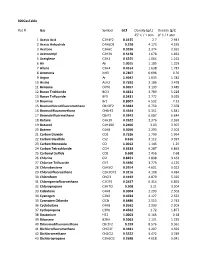
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Using SMILES Strings for the Description of Chemical Connectivity in the Crystallography Open Database
Quirós et al. J Cheminform (2018) 10:23 https://doi.org/10.1186/s13321-018-0279-6 RESEARCH ARTICLE Open Access Using SMILES strings for the description of chemical connectivity in the Crystallography Open Database Miguel Quirós1* , Saulius Gražulis2,3 , Saulė Girdzijauskaitė3, Andrius Merkys2 and Antanas Vaitkus2 Abstract Computer descriptions of chemical molecular connectivity are necessary for searching chemical databases and for predicting chemical properties from molecular structure. In this article, the ongoing work to describe the chemical connectivity of entries contained in the Crystallography Open Database (COD) in SMILES format is reported. This col- lection of SMILES is publicly available for chemical (substructure) search or for any other purpose on an open-access basis, as is the COD itself. The conventions that have been followed for the representation of compounds that do not ft into the valence bond theory are outlined for the most frequently found cases. The procedure for getting the SMILES out of the CIF fles starts with checking whether the atoms in the asymmetric unit are a chemically accept- able image of the compound. When they are not (molecule in a symmetry element, disorder, polymeric species,etc.), the previously published cif_molecule program is used to get such image in many cases. The program package Open Babel is then applied to get SMILES strings from the CIF fles (either those directly taken from the COD or those produced by cif_molecule when applicable). The results are then checked and/or fxed by a human editor, in a computer-aided task that at present still consumes a great deal of human time. -

Cyclobutadiene
View metadata, citation and similar papers at core.ac.uk brought to you by CORE Published in "Angewandte Chemie International Edition 45(40): 6616 - 6619, 20 provided by RERO DOC Digital Library which should be cited to refer to this work. Cyclobutadiene: The Antiaromatic Paradigm? Thomas Bally* Keywords: In memory of Satoru Masamune antiaromaticity · cyclobutadiene · thermochemistry (1928–2003) There is probably no other single widely accepted (and hardly disputed) London proposed in 1937 a very appeal- molecule that has fascinated experimen- that benzene, with its six cyclically ing model that accounts for these effects tal and theoretical chemists so consis- delocalized p electrons, represents the in terms of ring currents that are in- tently over the past 40 years as has paradigm of aromaticity. In view of the duced by the external magnetic field in cyclobutadiene (CBD). On average, famous Hckel 4n/4n + 2 electron rule, the system of cyclically delocalized 16 publications which deal, in one form one is tempted to rush to the conclusion p electrons.[5] or another, with the parent compound that CBD, with its four cyclically delo- There has been a vigorous debate in p C4H4 and 40 which deal with cyclo- calized electrons, is therefore the recent years about whether and to what butdienes in general have appeared in paradigm of antiaromaticity. extent these ring currents contribute to every year of this time span, and there However, this conclusion is not as the deshielding or shielding of protons are probably many more on derivatives straightforward as it might seem, be- attached to aromatic or antiaromatic and metal complexes. -

An Overview of Fullerene Chemistry
Bull. Mater. So., Vol. 20, No. 2, April 1997, pp. 141-230. © Printed in India. An overview of fullerene chemistry S SAMAL* and S K SAHOO Department of Chemistry, gavenshawCollege, Cuttack 753 003, India MS received 4 April 1996; revised 3 October 1996 Abstract. Fullerenes which were thought to be 'superaromatics' are actually 'super- alkenes'. Reactions in fuUerenesare varied, ranging from the addition of simple molecules like H 2 to large moleculeslike dendrimers.The synthesis,structure, characterization,along with simple reactions like halogenation, oxygenation, metalation,cydoaddition, polymeri- zation and dendrimer addition are discussed. Keywards. Fullerenes;C6o; C7o; characterization;reactions. 1. Introduction Fullerenes and their derivatives possess a number of potentially useful physical, biological and chemical properties. Our interest in the chemistry of fullerenes led us to present this review in which we have attempted to furnish an overall picture of the properties of these novel molecules. The scope of the review is however limited. The conclusion of the authors contributing to this rapidly growing field of research are accepted and presented in a concise manner highlighting the salient features of the original work. The technique developed by Kr/itschmer et al (1990) for preparing and isolating macroscopic quantities of C6o, 'a new form of carbon', opened the way for exploring the molecular and bulk properties of these novel species. Kroto et al (1985) reported the nature and chemical reactivity of C6o species produced during the nucleation of carbon plasma. C6o , which was found to be stable, was named buckminsterfullerene after Buckminster Fuller, American architect, engineer and constructor of geodesic dome.