Using SMILES Strings for the Description of Chemical Connectivity in the Crystallography Open Database
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

New Applications of Cyclobutadiene Cycloadditions: Diversity and Target Oriented Synthesis
New Applications of Cyclobutadiene Cycloadditions: Diversity and Target Oriented Synthesis Author: Jason Joseph Marineau Persistent link: http://hdl.handle.net/2345/1741 This work is posted on eScholarship@BC, Boston College University Libraries. Boston College Electronic Thesis or Dissertation, 2010 Copyright is held by the author, with all rights reserved, unless otherwise noted. Boston College The Graduate School of Arts and Sciences Department of Chemistry NEW APPLICATIONS OF CYCLOBUTADIENE CYCLOADDITIONS: DIVERSITY AND TARGET ORIENTED SYNTHESIS a dissertation by JASON JOSEPH MARINEAU submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy December 2010 © Copyright by JASON JOSEPH MARINEAU 2010 New Applications of Cyclobutadiene Cycloadditions: Diversity and Target Oriented Synthesis Jason J. Marineau Thesis Advisor: Professor Marc. L. Snapper Abstract Cyclobutadiene cycloadditions provide rapid access to rigid polycyclic systems with high strain energy and unusual molecular geometries. Further functionalization of these systems allows entry into unexplored chemical space. A tricarbonylcyclobutadiene iron complex on solid support enables exploration of these cycloadditions in a parallel format amenable to diversity oriented synthesis. Modeling of the cycloaddition transition states with density functional calculations provides a theoretical basis for analysis of the regioselectivity observed in generation of these substituted bicyclo[2.2.0]hexene derivatives. The high strain energy accessible in cyclobutadiene cycloadducts and their derivatives renders them useful synthons for access to medium-ring natural products through ring expansion. Torilin, a guaiane sesquiterpene isolated from extracts of the fruits of Torilis japonica, exhibits a range of biological activities including testosterone 5 -reductase inhibition, hKv1.5 channel blocking, hepatoprotective, anti-inflammatory and anti-cancer effects. -

CHEMISTRY 412/512 MIDTERM # 1 – Answer Key February 14, 2007 1
CHEMISTRY 412/512 MIDTERM # 1 – answer key February 14, 2007 1. (8 pts) Acyl cations ( RCO ) are key intermediates in Friedel – Crafts acylation reactions. They are stabilized by the presence of oxygen and this can be demonstrated through either resonance or MO analysis. a. Consider the formyl cation, shown below. Show how it is stabilized, using appropriate resonance structures. HCO formyl cation HCO HCO Acyl cations are stabilized by conjugation of one of the lone pairs at the oxygen center. b. Construct the orbital mixing diagram for the formyl cation, starting with group orbitals for CH and O-atom. Consider only first order mixing. Place the appropriate number of valence electrons and demonstrate how QMOT explains the stabilization of the cation. O HC The mixing diagram clearly shows the formation of TWO equivalent π-bonds, which is analogous to the resonance structure on the right, i.e. there is significant contribution by the O-atom. 2. (6 pts) Antiaromatic molecules avoid antiaromaticity by structural distortion. For example, cyclobutadiene is not square (and antiaromatic) but rectangular. Show, using π-group orbital analysis, that such distortion would indeed be stabilizing. square rectangular Solution: Instability of square cyclobutadiene is due to the presence of two degenerate, half-filled π-molecular orbitals (π2 and π3). In the rectangular form, qualitatively, the MOs remain the same, but the strength of some bonding/antibonding interactions is affected and it causes the degeneracy to be lifted. Focus on π2 and π3. Upon stretching, the antibonding interaction in π2 is reduced, while the same geometric distortion leads to reduction of the bonding interaction in π3. -

Indene CH Activation, Indole Π Vs. Nitrogen Lone-Pair Coordinati
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Organometallics Manuscript Author . Author Manuscript Author manuscript; available in PMC 2016 April 15. Published in final edited form as: Organometallics. 2007 ; 26(2): 281–287. doi:10.1021/om0606643. Reactions of Indene and Indoles with Platinum Methyl Cations: Indene C-H Activation, Indole π vs. Nitrogen Lone-Pair Coordination Travis J. Williams, Jay A. Labinger, and John E. Bercaw Arnold and Mabel Beckman Laboratories of Chemical Synthesis, California Institute of Technology, Pasadena, CA 91125 (U. S. A.) Abstract Reactions of indene and various substituted indoles with [(diimine)PtII(Me)(TFE)]+ cations have been studied (diimine = ArN = C(Me) − C(Me) = NAr; TFE = 2,2,2-trifluoroethanol). Indene displaces the TFE ligand from platinum to form a stable π coordination complex that, upon heating, undergoes C-H activation with first order kinetics, ΔH‡ = 29 kcal/mol, ΔS‡ = 10 eu, and a kinetic isotope effect of 1.1 at 60 °C. Indoles also initially form coordination complexes through the C2=C3 olefin, but these undergo rearrangement to the corresponding N-bound complexes. The relative rates of initial coordination and rearrangement are affected by excess acid or methyl substitution on indole. Introduction Selective C-H bond activation is a potentially valuable approach to synthetic problems in areas ranging from fuels and bulk chemicals to fine chemicals and pharmaceutical synthesis. 1 Studies of C-H activation in our laboratory have focused on models of the Shilov system,2 particularly [(diimine)PtII(Me)(solv)]+ (2, diimine = ArN = C(Me) − C(Me) = NAr; solv = 2,2,2-trifluoroethanol (TFE), H2O).3 These cations are capable of activating a variety of II carbon-hydrogen bonds.4 Cations 2 can be generated by protonolysis of (diimine)Pt Me2 species 1 in TFE with aqueous HBF43, 4ab or BX3 (X = C6F5,4cd F5), the latter producing H+ by boron coordination to TFE.4c In deuterated solvent 2 is formed as a mixture of two isotopologs. -

An Indicator of Triplet State Baird-Aromaticity
inorganics Article The Silacyclobutene Ring: An Indicator of Triplet State Baird-Aromaticity Rabia Ayub 1,2, Kjell Jorner 1,2 ID and Henrik Ottosson 1,2,* 1 Department of Chemistry—BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden; [email protected] (R.A.); [email protected] (K.J.) 2 Department of Chemistry-Ångström Laboratory Uppsala University, Box 523, SE-751 20 Uppsala, Sweden * Correspondence: [email protected]; Tel.: +46-18-4717476 Received: 23 October 2017; Accepted: 11 December 2017; Published: 15 December 2017 Abstract: Baird’s rule tells that the electron counts for aromaticity and antiaromaticity in the first ππ* triplet and singlet excited states (T1 and S1) are opposite to those in the ground state (S0). Our hypothesis is that a silacyclobutene (SCB) ring fused with a [4n]annulene will remain closed in the T1 state so as to retain T1 aromaticity of the annulene while it will ring-open when fused to a [4n + 2]annulene in order to alleviate T1 antiaromaticity. This feature should allow the SCB ring to function as an indicator for triplet state aromaticity. Quantum chemical calculations of energy and (anti)aromaticity changes along the reaction paths in the T1 state support our hypothesis. The SCB ring should indicate T1 aromaticity of [4n]annulenes by being photoinert except when fused to cyclobutadiene, where it ring-opens due to ring-strain relief. Keywords: Baird’s rule; computational chemistry; excited state aromaticity; Photostability 1. Introduction Baird showed in 1972 that the rules for aromaticity and antiaromaticity of annulenes are reversed in the lowest ππ* triplet state (T1) when compared to Hückel’s rule for the electronic ground state (S0)[1–3]. -

1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095
1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095-13-6 Justification: This compound is listed in Ohio Administrative Code 3745 - 114 - 01 because it fulfills one or more of the following criteria: substances that are known to be, or may reasonably be anticipated to be, carcinogenic, mutagenic, teratogenic, or neurotoxic, causes reproductive dysfunction, is acutely or chronically toxic, or causes the threat of adverse environmental effects through ambient concentrations, bioaccumulation, or atmospheric deposition. lndene is acutely toxic with effects on the upper respiratory system (mucous membranes), pulmonary irritation, liver and kidney effects. Molecular Weight (g/mol): 116.15 Synonyms: Indonaphthene U.S. EPA Carcinogenic Classification (IRIS): Not listed on IRIS. PBT: Not listed as persistent, bioaccumulative and toxic. NTP: Not listed by the National Toxicology Program. HAP: Not listed as a hazardous air pollutant (HAP) by U.S. EPA. 112r: Not listed under Section 112(r) of the Clean Air Act. ACGIH: TLV: 10 ppm or 47,505 µg/m3. Critical effects include upper respiratory irritation or damage, pulmonary irritation, and liver and kidney effects. HSDB: Listed in the Hazardous Substances Data Bank. Inhalation of indene vapors is expected to cause irritation of mucous membranes. International IARC: Not listed by International Agency for Research on Cancer (IARC). ATSDR (MRL): Not listed by ATSDR. DataSheet Indene.wpd 2/2 Reference Material 1. American Conference of Governmental Industrial Hygienists (ACGIH) 2006. TLVs and BEIs: -
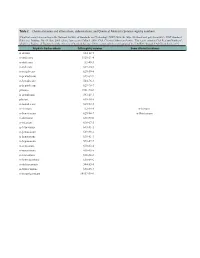
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Cyclobutadiene
View metadata, citation and similar papers at core.ac.uk brought to you by CORE Published in "Angewandte Chemie International Edition 45(40): 6616 - 6619, 20 provided by RERO DOC Digital Library which should be cited to refer to this work. Cyclobutadiene: The Antiaromatic Paradigm? Thomas Bally* Keywords: In memory of Satoru Masamune antiaromaticity · cyclobutadiene · thermochemistry (1928–2003) There is probably no other single widely accepted (and hardly disputed) London proposed in 1937 a very appeal- molecule that has fascinated experimen- that benzene, with its six cyclically ing model that accounts for these effects tal and theoretical chemists so consis- delocalized p electrons, represents the in terms of ring currents that are in- tently over the past 40 years as has paradigm of aromaticity. In view of the duced by the external magnetic field in cyclobutadiene (CBD). On average, famous Hckel 4n/4n + 2 electron rule, the system of cyclically delocalized 16 publications which deal, in one form one is tempted to rush to the conclusion p electrons.[5] or another, with the parent compound that CBD, with its four cyclically delo- There has been a vigorous debate in p C4H4 and 40 which deal with cyclo- calized electrons, is therefore the recent years about whether and to what butdienes in general have appeared in paradigm of antiaromaticity. extent these ring currents contribute to every year of this time span, and there However, this conclusion is not as the deshielding or shielding of protons are probably many more on derivatives straightforward as it might seem, be- attached to aromatic or antiaromatic and metal complexes. -

The Relation Between Ring Currents and Aromatic Stabilization Energies for Assessing the Degree of Aromaticity
The Relation Between Ring Currents and Aromatic Stabilization Energies for Assessing the Degree of Aromaticity Chandan Kumar,y Heike Fliegl,∗,y and Dage Sundholm∗,z yCentre for Theoretical and Computational Chemistry, Department of Chemistry, University of Oslo, P.O.Box 1033, N-1315 Blindern, Norway zDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A. I. Virtanens Plats 1), FIN-00014, Finland E-mail: heike.fl[email protected]; [email protected].fi 1 Abstract Magnetically induced ring-current strength susceptibilities and nucleus independent chemical shifts (NICS) have been studied for 15 single-ring aromatic, antiaromatic and nonaromatic molecules. The current densities have been calculated at the density func- tional theory (DFT), Hartree-Fock (HF), and second-order Møller-Plesset perturbation theory (MP2) levels using the gauge-including magnetically induced current method (GIMIC). The ring-current strength susceptibilities have been obtained by numerical integration of the current density flowing around the molecular ring. The calculated ring-current strength susceptibilities are almost independent of the level of theory. The relative degree of aromaticity deduced from the magnetic properties has been compared with the ones deduced from hydrogenation enthalpies that are considered to be propor- tional to aromatic stabilization energies (ASE). For the studied single-ring molecules, GIMIC, NICS and ASE calculations yield similar trends. The study shows that there is a linear correlation between the magnetic and energetic criteria of aromaticity. The largest uncertainty originates from the accuracy of the energy data, because they are much more dependent on the employed computational level than the calculated mag- netic properties. Thus, ring-current strength susceptibilities can be used for assessing the degree of aromaticity. -

Highly Unsaturated Binuclear Butadiene Iron Carbonyls: Quintet Spin States, Perpendicular Structures, Agostic Hydrogen Atoms, and Iron-Iron Multiple Bonds
Int. J. Mol. Sci. 2011, 12, 2216-2231; doi:10.3390/ijms12042216 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 http://www.mdpi.com/journal/ijms Article Highly Unsaturated Binuclear Butadiene Iron Carbonyls: Quintet Spin States, Perpendicular Structures, Agostic Hydrogen Atoms, and Iron-Iron Multiple Bonds Yi Zeng 1, Shijian Wang 1, Hao Feng 1,*, Yaoming Xie 2 and R. Bruce King 2,* 1 School of Physics and Chemistry, Research Center for Advanced Computation, Xihua University, Chengdu, China; E-Mails: [email protected] (Y.Z.); [email protected] (S.W.) 2 Department of Chemistry and Center for Computational Chemistry; University of Georgia, Athens, GA 30602, USA; E-Mail: [email protected] * Authors to whom correspondence should be addressed; E-Mails: [email protected] (H.F.); [email protected] (R.B.K.); Tel.: +86-28-87727663 (H.F.); +1-706-542-1901 (R.B.K.); Fax: +1-706-542-9454 (R.B.K.). Received: 22 February 2011; in revised form: 8 March 2011 / Accepted: 14 March 2011 / Published: 30 March 2011 Abstract: The highly unsaturated binuclear butadiene iron carbonyls (C4H6)2Fe2(CO)n (n = 2, 1) have been examined using density functional theory. For (C4H6)2Fe2(CO)n (n = 2, 1), both coaxial and perpendicular structures are found. The global minima of (C4H6)2Fe2(CO)n (n = 2, 1) are the perpendicular structures 2Q-1 and 1Q-1, respectively, with 17- and 15-electron configurations for the iron atoms leading to quintet spin states. The Fe=Fe distance of 2.361 Å (M06-L) in the (C4H6)2Fe2(CO)2 structure 2Q-1 suggests a formal double bond. -

Carcinogens CAS DOT SHHC Sources Number Chemical Name
2010 Right to Know Special Health Hazardous Substance List Substance Common Name Carcinogens CAS DOT SHHC Sources Number Chemical Name 3140 # ACEPHATE 30560-19-1 2783 CA 3 6 8 17 18 PHOSPHORAMIDOTHIOIC ACID, ACETYL-, O,S-DIMETHYL ESTER 0001 # ACETALDEHYDE 75-07-0 1089 CA MU TE F4 1 2 3 4 5 6 7 8 R2 15 17 18 20 21 ACETALDEHYDE 22 2890 # ACETAMIDE 60-35-5 3077 CA 3 6 7 17 18 20 ACETAMIDE 0010 # 2-ACETYLAMINOFLUORENE 53-96-3 CA MU 1 4 5 6 18 20 21 ACETAMIDE, N-9H-FLUOREN-2-YL- 0022 # ACRYLAMIDE 79-06-1 2074 CA R2 1 2 3 4 5 6 7 8 15 17 18 19 20 2-PROPENAMIDE 21 0024 # ACRYLONITRILE 107-13-1 1093 CA TE F3 R2 1 2 3 4 5 6 7 8 14 15 17 18 19 2-PROPENENITRILE 20 21 22 3142 # AF- 2 3688-53-7 CA 7 2-FURANACETAMIDE, .alpha.-[(5-NITRO-2-FURANYL)METHYLENE]- 0029 # AFLATOXINS 1402-68-2 CA MU TE 5 7 AFLATOXINS 0033 # ALDRIN 309-00-2 2761 CA TE 1 2 3 4 6 7 8 14 17 18 19 20 21 1,4:5,8-DIMETHANONAPHTHALENE, 1,2,3,4,10,10-HEXACHLORO-1,4,4a,5,8,8aHEXAHYDRO(1R,4S,4aS,5S,8R,8aR)-rel- Page 1 of 55 2010 Right to Know Special Health Hazardous Substance List Substance Common Name Carcinogens CAS DOT SHHC Sources Number Chemical Name 0039 # ALLYL CHLORIDE 107-05-1 1100 CA F3 1 2 3 4 6 7 8 15 17 18 20 1-PROPENE, 3-CHLORO- 0069 # 2-AMINOANTHRAQUINONE 117-79-3 CA MU 5 6 7 18 9,10-ANTHRACENEDIONE, 2-AMINO- 4012 # 1-AMINO-2,4-DIBROMOANTHRAQUINONE 81-49-2 CA 5 9,10-ANTHRACENEDIONE, 1-AMINO-2,4-DIBROMO- 0072 # 4-AMINODIPHENYL 92-67-1 CA MU 1 2 4 5 6 7 18 20 [1,1'-BIPHENYL]-4-AMINE 0076 # 1-AMINO-2-METHYLANTHRAQUINONE 82-28-0 CA 5 6 7 18 9,10-ANTHRACENEDIONE, 1-AMINO-2-METHYL- -

Fluorinated Butatrienes
Fluorinated Butatrienes Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) eingereicht im Fachbereich Biologie, Chemie, Pharmazie der Freien Universität Berlin vorgelegt von Dipl.-Chem. Christian Ehm aus Berlin Berlin, April 2010 1. Gutachter: Prof. Dr. Dieter Lentz 2. Gutachter: Prof. Dr. Beate Paulus Disputation am 28.6.2010 I Acknowledgements It would not have been possible to write this doctoral thesis without the help and support of the kind people around me, to only some of whom it is possible to give particular mention here. First and foremost I would like to thank my principal supervisor, Professor Dieter Lentz, for the opportunity of doing research in his group. Without his continuous support and encouragement this thesis would not be in the present state. I highly appreciate that Professor Beate Paulus has agreed to be co-referee of my thesis. I would like to cordially thank Lada for her love and patience as well as her interest in my research. Special thanks to my family for their continuous support and love. I would like to thank Mike Roland, Sten Dathe and Sven Wünsche for their friendship and the fun we have had every Sunday evening. Special thanks to Sebastian Freitag, Boris Bolsinger and Frederic Heinrich for their friendship. They deserve much gratefulness for keeping me on the right way. I would like to thank all my colleagues at the Institut für Chemie und Biochemie, Abteilung Anorganische Chemie. In particular I want to thank all members of the Lentz group, Thomas Hügle, Moritz Kühnel, Dr. Floris Akkerman, Dr. -

I. the Low Temperature Photochemistry of Iron Pentacarbonyl and Disubstituted Acetylenes, II
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1971 I. The low temperature photochemistry of iron pentacarbonyl and disubstituted acetylenes, II. The X-ray structure determination of trans-6,8-dibromo-1,2,3,4,4a,9a- hexahydro-4a,9-dimethylcarbazole Allen Bloom Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Bloom, Allen, "I. The low temperature photochemistry of iron pentacarbonyl and disubstituted acetylenes, II. The -rX ay structure determination of trans-6,8-dibromo-1,2,3,4,4a,9a-hexahydro-4a,9-dimethylcarbazole " (1971). Retrospective Theses and Dissertations. 4943. https://lib.dr.iastate.edu/rtd/4943 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. 72-5178 BLOOM, Allen, 1943- Ï. THE LOW TEMPERATURE PHOTOCHEMISTRY OF IRON PENTACARBONYL AND DISUBSTITUTED ACETYLENES. II. THE X-RAY STRUCTURE DETERMINATION OF trans- 6,8,-DIBROMO-l,2,3,4,4a,9a-HEXAHYDR0-4a,9- DIMETHYLCARBAZOLE. Iowa State University, Ph.D., 1971 Chemistry, organic University Microfilms, A XEROX Company, Ann Arbor, Michigan THIS DISSERTATION HAS BEEN MICROFLIMED EXACTLY AS RECEIVED I. The low temperature photochemistry of iron pentacarbonyl and disubstituted acetylenes II. The X-ray structure determination of tranB-6,8- dibromo-1,2,3,4,4a,9a-hexahydro-4a,9-dimethylcarbazole By Allen Bloom  Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved: Signature was redacted for privacy.