Indene CH Activation, Indole Π Vs. Nitrogen Lone-Pair Coordinati
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095
1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095-13-6 Justification: This compound is listed in Ohio Administrative Code 3745 - 114 - 01 because it fulfills one or more of the following criteria: substances that are known to be, or may reasonably be anticipated to be, carcinogenic, mutagenic, teratogenic, or neurotoxic, causes reproductive dysfunction, is acutely or chronically toxic, or causes the threat of adverse environmental effects through ambient concentrations, bioaccumulation, or atmospheric deposition. lndene is acutely toxic with effects on the upper respiratory system (mucous membranes), pulmonary irritation, liver and kidney effects. Molecular Weight (g/mol): 116.15 Synonyms: Indonaphthene U.S. EPA Carcinogenic Classification (IRIS): Not listed on IRIS. PBT: Not listed as persistent, bioaccumulative and toxic. NTP: Not listed by the National Toxicology Program. HAP: Not listed as a hazardous air pollutant (HAP) by U.S. EPA. 112r: Not listed under Section 112(r) of the Clean Air Act. ACGIH: TLV: 10 ppm or 47,505 µg/m3. Critical effects include upper respiratory irritation or damage, pulmonary irritation, and liver and kidney effects. HSDB: Listed in the Hazardous Substances Data Bank. Inhalation of indene vapors is expected to cause irritation of mucous membranes. International IARC: Not listed by International Agency for Research on Cancer (IARC). ATSDR (MRL): Not listed by ATSDR. DataSheet Indene.wpd 2/2 Reference Material 1. American Conference of Governmental Industrial Hygienists (ACGIH) 2006. TLVs and BEIs: -

Using SMILES Strings for the Description of Chemical Connectivity in the Crystallography Open Database
Quirós et al. J Cheminform (2018) 10:23 https://doi.org/10.1186/s13321-018-0279-6 RESEARCH ARTICLE Open Access Using SMILES strings for the description of chemical connectivity in the Crystallography Open Database Miguel Quirós1* , Saulius Gražulis2,3 , Saulė Girdzijauskaitė3, Andrius Merkys2 and Antanas Vaitkus2 Abstract Computer descriptions of chemical molecular connectivity are necessary for searching chemical databases and for predicting chemical properties from molecular structure. In this article, the ongoing work to describe the chemical connectivity of entries contained in the Crystallography Open Database (COD) in SMILES format is reported. This col- lection of SMILES is publicly available for chemical (substructure) search or for any other purpose on an open-access basis, as is the COD itself. The conventions that have been followed for the representation of compounds that do not ft into the valence bond theory are outlined for the most frequently found cases. The procedure for getting the SMILES out of the CIF fles starts with checking whether the atoms in the asymmetric unit are a chemically accept- able image of the compound. When they are not (molecule in a symmetry element, disorder, polymeric species,etc.), the previously published cif_molecule program is used to get such image in many cases. The program package Open Babel is then applied to get SMILES strings from the CIF fles (either those directly taken from the COD or those produced by cif_molecule when applicable). The results are then checked and/or fxed by a human editor, in a computer-aided task that at present still consumes a great deal of human time. -
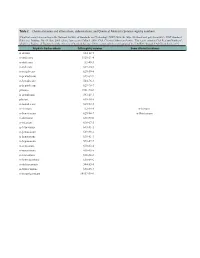
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Carcinogens CAS DOT SHHC Sources Number Chemical Name
2010 Right to Know Special Health Hazardous Substance List Substance Common Name Carcinogens CAS DOT SHHC Sources Number Chemical Name 3140 # ACEPHATE 30560-19-1 2783 CA 3 6 8 17 18 PHOSPHORAMIDOTHIOIC ACID, ACETYL-, O,S-DIMETHYL ESTER 0001 # ACETALDEHYDE 75-07-0 1089 CA MU TE F4 1 2 3 4 5 6 7 8 R2 15 17 18 20 21 ACETALDEHYDE 22 2890 # ACETAMIDE 60-35-5 3077 CA 3 6 7 17 18 20 ACETAMIDE 0010 # 2-ACETYLAMINOFLUORENE 53-96-3 CA MU 1 4 5 6 18 20 21 ACETAMIDE, N-9H-FLUOREN-2-YL- 0022 # ACRYLAMIDE 79-06-1 2074 CA R2 1 2 3 4 5 6 7 8 15 17 18 19 20 2-PROPENAMIDE 21 0024 # ACRYLONITRILE 107-13-1 1093 CA TE F3 R2 1 2 3 4 5 6 7 8 14 15 17 18 19 2-PROPENENITRILE 20 21 22 3142 # AF- 2 3688-53-7 CA 7 2-FURANACETAMIDE, .alpha.-[(5-NITRO-2-FURANYL)METHYLENE]- 0029 # AFLATOXINS 1402-68-2 CA MU TE 5 7 AFLATOXINS 0033 # ALDRIN 309-00-2 2761 CA TE 1 2 3 4 6 7 8 14 17 18 19 20 21 1,4:5,8-DIMETHANONAPHTHALENE, 1,2,3,4,10,10-HEXACHLORO-1,4,4a,5,8,8aHEXAHYDRO(1R,4S,4aS,5S,8R,8aR)-rel- Page 1 of 55 2010 Right to Know Special Health Hazardous Substance List Substance Common Name Carcinogens CAS DOT SHHC Sources Number Chemical Name 0039 # ALLYL CHLORIDE 107-05-1 1100 CA F3 1 2 3 4 6 7 8 15 17 18 20 1-PROPENE, 3-CHLORO- 0069 # 2-AMINOANTHRAQUINONE 117-79-3 CA MU 5 6 7 18 9,10-ANTHRACENEDIONE, 2-AMINO- 4012 # 1-AMINO-2,4-DIBROMOANTHRAQUINONE 81-49-2 CA 5 9,10-ANTHRACENEDIONE, 1-AMINO-2,4-DIBROMO- 0072 # 4-AMINODIPHENYL 92-67-1 CA MU 1 2 4 5 6 7 18 20 [1,1'-BIPHENYL]-4-AMINE 0076 # 1-AMINO-2-METHYLANTHRAQUINONE 82-28-0 CA 5 6 7 18 9,10-ANTHRACENEDIONE, 1-AMINO-2-METHYL- -

Coal Tar Creosote
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization. Concise International Chemical Assessment Document 62 COAL TAR CREOSOTE Please note that the layout and pagination of this pdf file are not identical to the document being printed First draft prepared by Drs Christine Melber, Janet Kielhorn, and Inge Mangelsdorf, Fraunhofer Institute of Toxicology and Experimental Medicine, Hanover, Germany Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 2004 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. -

Chlordane and Heptachlor
CHLORDANE AND HEPTACHLOR Chlordane and heptachlor were considered together because of their close structural similarity and because technical-grade products each contain about 10–20% of the other compound. These substances were considered by previous working groups, in 1978 (IARC, 1979), 1987 (IARC, 1987) and 1990 (IARC, 1991). Since that time, new data have become available, and these have been incorporated into the monograph and taken into consideration in the present evaluation. 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Synonyms, structural and molecular data Chemical Abstract Services Registry numbers, names and synonyms of chlordane and heptachlor and its epoxide are given in Table 1. Cl Cl Cl Cl Cl Cl Cl Cl C10H6Cl8 Chlordane Relative molecular mass: 409.8 Cl Cl Cl Cl Cl Cl Cl C10H5Cl7 Heptachlor Relative molecular mass: 373.5 –411– 412 IARC MONOGRAPHS VOLUME 79 Table 1. Chemical Abstract Services Registry numbers, names and synonyms of chlordane, heptachlor and its epoxide Name CAS Reg. Nosa Chem. Abstr. namesb and synonyms Chlordane 57-74-9 ENT 9932; 1,2,4,5,6,7,8,8-octachloro-2,3,3a,4,7,7a- (39400-80-1); hexahydro-4,7-methano-1H-indene; 1,2,4,5,6,7,8,8- 53637-13-1) octachloro-2,3,3a,4,7,7a-hexahydro-4,7-methano- indene (IUPAC); octachloro-4,7-methanotetrahydro- indane; 1,2,4,5,6,7,8,8-octachloro-3a,4,7,7a-tetra- hydro-4,7-methanoindan; OMS 1437 Technical-grade 12789-03-6 chlordane cis-Chlordane 5103-71-9 α-Chlordan; α-chlordane; cis-chlordan; (152322-29-7; (1α,2α,3aα,4β,7β,7aα)-1,2,4,5,6,7,8,8-octachloro- 22212-52-8; -

EPA's Hazardous Waste Listing
Hazardous Waste Listings A User-Friendly Reference Document September 2012 Table of Contents Introduction ..................................................................................................................................... 3 Overview of the Hazardous Waste Identification Process .............................................................. 5 Lists of Hazardous Wastes .............................................................................................................. 5 Summary Chart ............................................................................................................................... 8 General Hazardous Waste Listing Resources ................................................................................. 9 § 261.11 Criteria for listing hazardous waste. .............................................................................. 11 Subpart D-List of Hazardous Wastes ............................................................................................ 12 § 261.31 Hazardous wastes from non-specific sources. ............................................................... 13 Spent solvent wastes (F001 – F005) ......................................................................................... 13 Wastes from electroplating and other metal finishing operations (F006 - F012, and F019) ... 18 Dioxin bearing wastes (F020 - F023, and F026 – F028) .......................................................... 22 Wastes from production of certain chlorinated aliphatic hydrocarbons (F024 -

ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability Of
ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability of Coastal Plain Groundwater to Flood Water Intrusion using High Resolution Mass Spectrometry. (Under the direction of Dr. Elizabeth Guthrie Nichols). Communities in the North Carolina Coastal Plain (NCCP) depend on safe and reliable groundwater for private well use, agriculture, industry, and livelihoods. Although storm intensity and frequency are predicted to increase in coastal areas, the risk of surficial and confined aquifer contamination from extreme storms is not understood. In September 2018, Hurricane Florence caused extensive flooding across the NCCP for several weeks. The North Carolina Department of Environmental Quality (NCDEQ) Groundwater Management Branch had just completed sampling of some wells in their monitoring network when Hurricane Florence made landfall. NCDEQ returned to these wells, particularly those flooded by Hurricane Florence, for post-flood sampling. These groundwater samples were analyzed by NCDEQ for regulated semi-volatile organics with few to any detections of regulated organic contaminants. NCDEQ provided NC State the same sample extracts for analysis by high resolution mass spectrometry (HRMS). This research reports on the non-targeted and suspect-screening HRMS analyses of groundwater from nested monitoring wells. Some monitoring well sites experienced flooding during the study period, and some did not. The goal of this research was to advance our understanding of coastal aquifer susceptibility to flooding by producing the first comprehensive organic chemical profiles of coastal aquifers and by determining if aquifers have distinct organic chemical profiles that change after flooding events. This study used HRMS analyses to produce the first comprehensive organic chemical profiles of 11 aquifers in the coastal plain. -

A Chemical Kinetic Modeling Study of Indene Pyrolysis
A chemical kinetic modeling study of indene pyrolysis Item Type Article Authors Jin, Hanfeng; Xing, Lili; Hao, Junyu; Yang, Jiuzhong; Zhang, Yan; Cao, Chuangchuang; Pan, Yang; Farooq, Aamir Citation Jin H, Xing L, Hao J, Yang J, Zhang Y, et al. (2019) A chemical kinetic modeling study of indene pyrolysis. Combustion and Flame 206: 1–20. Available: http://dx.doi.org/10.1016/ j.combustflame.2019.04.040. DOI 10.1016/j.combustflame.2019.04.040 Publisher Elsevier BV Journal Combustion and Flame Download date 02/10/2021 19:12:45 Link to Item http://hdl.handle.net/10754/653053 1 A chemical kinetic modeling study of indene pyrolysis 2 Hanfeng Jin1, Lili Xing2*, Junyu Hao1, Jiuzhong Yang3, Yan Zhang4, ChuangChuang Cao4, Yang 3 1* 3 Pan , Aamir Farooq 4 1Clean Combustion Research Centre, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia 5 2Energy and Power Engineering Institute, Henan University of Science and Technology, Luoyang, Henan 471003, China 6 3National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China 7 4Key Laboratory for Power Machinery and Engineering of MOE, Shanghai Jiao Tong University, Shanghai 200240, China 8 9 Abstract 10 An improved polycyclic aromatic hydrocarbon (PAH) model is developed to predict the 11 decomposition of indene and the formation of large PAHs under pyrolytic conditions. This model is 12 developed based on experimental study of pyrolytic kinetics of indene in a flow reactor at low and 13 atmospheric pressures (30 and 760 Torr) by using synchrotron vacuum ultraviolet photoionization 14 mass spectrometry (SVUV-PIMS). -

Indene and Naphthalene Derivatives
Patentamt 0 1 99 393 JEuropaischesEuropean Patent Office Publication number: B1 Office europeen des brevets EUROPEAN PATENT SPECIFICATION 07 C 83/10, C 07 C 123/00, (45) Date of publication of patent specification: 07.12.88 Intel.4: C C 07 C 133/12, C 07 C 135/00, ® Application number: 86200495.9 A 61 K 31/155 ® Date of filing: 24.03.86 Indene and naphthalene derivatives. (§j) Priority: 02.04.85 GB 8508588 Proprietor: AKZON.V. Velperweg 76 NL-6824 BM Arnhem (NL) Date of publication of application: 29.10.86 Bulletin 86/44 Inventor: Logan, Robert Thomas "Leven"1StaikHHI Scotland (GB) Publication of the grant of the patent: Lanark ML11 9PW Lanarkshire 07.12.88 Bulletin 88/49 Inventor: Redpath, James 5 Etive Crescent Bishopbriggs Glasgow Scotland (GB) Gibson Designated Contracting States: Inventor: Roy, Robert BECHDEFRGBITLINLSE 63 Kenshaw Avenue Larkhall ML9 1PW Lanarkshire Scotland (GB) cited: References G.M. et al EP-A-0 030 749 Representative: Hermans, Franciscus FR-A-2 504 526 Postbus 20 US-A-3130 232 NL-5340 BH Oss (NL) US-A-4013 682 CO US-A-4 076 726 CO o CO The file contains technical information filed and o submitted after the application was not included in this specification Note- Within nine months from the publication of the mention of the grant of the European patent, any person may Notice of shall give notice to the European Patent Office of opposition to the European patent granted. opposition be deemed to have been filed until the opposition fee has been Q. -

Detailed Experimental and Kinetic Modeling Study of Toluene/C2 Pyrolysis in a Single-Pulse Shock Tube
Combustion and Flame 226 (2021) 129–142 Contents lists available at ScienceDirect Combustion and Flame journal homepage: www.elsevier.com/locate/combustflame Detailed experimental and kinetic modeling study of toluene/C 2 pyrolysis in a single-pulse shock tube ∗ ∗ Wenyu Sun a, , Alaa Hamadi a, Said Abid a,b, Nabiha Chaumeix a, Andrea Comandini a, a CNRS-INSIS, I.C.A.R.E., 1C, Avenue de la recherche scientifique, 45071 Orléans cedex 2, France b Université d’Orléans, 6 Avenue du Parc Floral, 45100 Orléans, France a r t i c l e i n f o a b s t r a c t Article history: A combined experimental and kinetic modeling study is carried out to explore the influences of acetylene Received 17 September 2020 and ethylene addition on the species formation from toluene pyrolysis. Experiments are conducted sepa- Revised 27 November 2020 rately with four different argon-diluted binary toluene/C 2 mixtures in a single pulse shock tube at a nom- Accepted 27 November 2020 inal pressure of 20 bar over a temperature range of 1150 −1650 K. All the experimental mixtures contain about 100 ppm toluene and different amounts of C 2 fuels (50, 216, 459 ppm acetylene and 516 ppm ethy- Keywords: lene). Species concentrations as a function of the temperature are probed from the post-shock gas mix- Toluene tures and analyzed through the gas chromatography/gas chromatography-mass spectrometry techniques. Acetylene A kinetic model is developed, which successfully predicts the absolute species concentration measure- Ethylene ments as well as the changes brought by the varied fuel compositions. -

The Chemistry of Homophthalic Acid
DEVELOPMENT OF NEW SYNTHETIC METHODOLOGIES FOR THE SYNTHESIS OF UNUSUAL ISOCOUMARIN AND INDOLE DERIVATIVES: THE CHEMISTRY OF HOMOPHTHALIC ACID A THESIS SUBMITTED TO THE GRADUATE SCHOOL OF NATURAL AND APPLIED SCIENCES OF MIDDLE EAST TECHNICAL UNIVERSITY BY SEVİL ÖZCAN IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGRE OF DOCTOR OF PHILOSOPHY IN DEPARTMENT OF CHEMISTRY JANUARY 2007 Approval of the Graduate School of Natural and Applied Sciences Prof. Dr. Canan Özgen Director I certify that this thesis satisfies all the requirements as a thesis for the degree of Doctor of Philosophy. Prof. Dr. Ahmet Önal Head of Department This is to certify that we have read this thesis and that in our opinion it is fully adequate, in scope and quality, as a thesis for the degree of Doctor of Philosophy. Prof. Dr. Metin Balcı Supervisor Examining Committee Members Prof. Dr. Canan Ünaleroğlu (Hacettepe UNV, CHEM) Prof. Dr. Metin Balcı(METU, CHEM) Prof. Dr. Basri Atasoy (GAZİ UNV., CHEM EDUC.) Prof. Dr. Bekir Peynircioğlu (METU, CHEM) Prof. Dr. Metin Zora (METU, CHEM.) I hereby declare that all information in this document has been obtained and presented in accordance with academic rules and ethical conduct. I also declare that, as required by these rules and conduct, I have fully cited and referenced all material and results that are not original to this work. Name, Last Name: SEVİL ÖZCAN Signature : iii ABSTRACT DEVELOPMENT OF NEW SYNTHETIC METHODOLOGIES FOR THE SYNTHESIS OF UNUSUAL ISOCOUMARIN AND INDOLE DERIVATIVES: THE CHEMISTRY OF HOMOPHTHALIC ACID ÖZCAN, Sevil Ph. D., Department of Chemistry Supervisor: Prof. Dr.