Photoelectron Spectroscopy of Heterocycles. Indene Analogs H
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

68 3 1 5"! Patent Request: Standard Patent/Patent of Addition
Λ _ ___ _ KWU/lXHJIMI AUSTRALIA Patents Act 1990 68 3 1 5"! PATENT REQUEST: STANDARD PATENT/PATENT OF ADDITION We, being the persons identified below as the Applicant, request the grant of a patent to the person identified below as the Nominated Person, for an invention described in the accompanying standard complete specification. ,· Full application details follow. [71] Applicant: ADIR ET COMPAGNIE Address: 1 RUE cXrLE HEBERT, F-92415 COURBEVOIE CEDEX, FRANCE [70] Nominated Person: ADIR ET COMPAGNIE Address: 1 RUE CARLE HEBERT, F-92415 COURBEVOIE CEDEX, FRANCE [54] Invent»·* Title: NOVEL N-PYRIDYL CARBOXAMIDES AND DERIVATIVES, PROCESSES FOR THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS WHICH CONTAIN THEM Name(s) of actual inventor(s): JEAN-MICHEL ROBERT, ODILE RIDEAU, SYLVIE ROBERT-PIESSARD, JACQUELINE COURANT, GUILLAUME LE BAUT, DANIEL-HENRI CAIGNARD, PIERRE RENARD and GERARD ADAM Address for service in Australia: c/o WATERMARK PATENT & TRADEMARK ATTORNEYS, of 290 Burwood Road, Hawthorn, Victoria 3122, Australia Attorney Code: WM :,.··. BASIC CONVENTION APPLICATION(S) DETAILS .... [31] Application Number [33] Country Country [32] Date of Application : Code 9406412 FRANCE FR 27 MAY 1994 Basic Applicants): ADIR ET COMPAGNIE • · · · • · · *· Di awing number recommended to accompany the abstract ............................... By our Patent Attorneys, WATERMARK PATENT & TRADEMARK ATTORNEYS ...CM&/.VV2...... ....... DATED this 25th day of May 1995,. Carolyn J, Harris Registered Patent Attorney i P/00/008b 12/11/91 Section 29 (η Regulation 3.1 (2) AUSTRALIA Patents Act 1990 NOTICE OF ENTITLEMENT We, ADIR ET COMPAGNIE of, 1 Rue Carle Hebert, F-92415 Courbevoie Cedex, France, being the applicant in respect of Application No. -

Indene CH Activation, Indole Π Vs. Nitrogen Lone-Pair Coordinati
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Organometallics Manuscript Author . Author Manuscript Author manuscript; available in PMC 2016 April 15. Published in final edited form as: Organometallics. 2007 ; 26(2): 281–287. doi:10.1021/om0606643. Reactions of Indene and Indoles with Platinum Methyl Cations: Indene C-H Activation, Indole π vs. Nitrogen Lone-Pair Coordination Travis J. Williams, Jay A. Labinger, and John E. Bercaw Arnold and Mabel Beckman Laboratories of Chemical Synthesis, California Institute of Technology, Pasadena, CA 91125 (U. S. A.) Abstract Reactions of indene and various substituted indoles with [(diimine)PtII(Me)(TFE)]+ cations have been studied (diimine = ArN = C(Me) − C(Me) = NAr; TFE = 2,2,2-trifluoroethanol). Indene displaces the TFE ligand from platinum to form a stable π coordination complex that, upon heating, undergoes C-H activation with first order kinetics, ΔH‡ = 29 kcal/mol, ΔS‡ = 10 eu, and a kinetic isotope effect of 1.1 at 60 °C. Indoles also initially form coordination complexes through the C2=C3 olefin, but these undergo rearrangement to the corresponding N-bound complexes. The relative rates of initial coordination and rearrangement are affected by excess acid or methyl substitution on indole. Introduction Selective C-H bond activation is a potentially valuable approach to synthetic problems in areas ranging from fuels and bulk chemicals to fine chemicals and pharmaceutical synthesis. 1 Studies of C-H activation in our laboratory have focused on models of the Shilov system,2 particularly [(diimine)PtII(Me)(solv)]+ (2, diimine = ArN = C(Me) − C(Me) = NAr; solv = 2,2,2-trifluoroethanol (TFE), H2O).3 These cations are capable of activating a variety of II carbon-hydrogen bonds.4 Cations 2 can be generated by protonolysis of (diimine)Pt Me2 species 1 in TFE with aqueous HBF43, 4ab or BX3 (X = C6F5,4cd F5), the latter producing H+ by boron coordination to TFE.4c In deuterated solvent 2 is formed as a mixture of two isotopologs. -

Dibenzothiophene/Oxide and Quinoxaline/Pyrazine Derivatives Serving As Electron-Transport Materials**
FULL PAPER DOI: 10.1002/adfm.200500823 Dibenzothiophene/Oxide and Quinoxaline/Pyrazine Derivatives Serving as Electron-Transport Materials** By Tai-Hsiang Huang, Wha-Tzong Whang,* Jiun Yi Shen, Yuh-Sheng Wen, Jiann T. Lin,* Tung-Huei Ke, Li-Yin Chen, and Chung-Chih Wu* A series of 2,8-disubstituted dibenzothiophene and 2,8-disubstituted dibenzothiophene-S,S-dioxide derivatives containing quinoxaline and pyrazine moieties are synthesized via three key steps: i) palladium-catalyzed Sonogashira coupling reaction to form dialkynes; ii) conversion of the dialkynes to diones; and iii) condensation of the diones with diamines. Single-crystal characterization of 2,8-di(6,7-dimethyl-3-phenyl-2-quinoxalinyl)-5H-5k6-dibenzo[b,d]thiophene-5,5-dione indicates a triclinic crystal structure with space group P1 and a non-coplanar structure. These new materials are amorphous, with glass-transition temperatures ranging from 132 to 194 °C. The compounds (Cpd) exhibit high electron mobilities and serve as effective elec- tron-transport materials for organic light-emitting devices. Double-layer devices are fabricated with the structure indium tin oxide (ITO)/Qn/Cpd/LiF/Al, where yellow-emitting 2,3-bis[4-(N-phenyl-9-ethyl-3-carbazolylamino)phenyl]quinoxaline (Qn) serves as the emitting layer. An external quantum efficiency of 1.41 %, a power efficiency of 4.94 lm W–1, and a current efficien- cy of 1.62 cd A–1 are achieved at a current density of 100 mA cm–2. 1. Introduction efficiency, brightness, and durability.[4] In contrast, reports of the use of small molecules as electron-transporting materials [5] Organic and polymer light-emitting diodes (OLEDs and are still rare in the literature. -

1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095
1/2 Toxic Compound Data Sheet Name: Indene CAS Number: 00095-13-6 Justification: This compound is listed in Ohio Administrative Code 3745 - 114 - 01 because it fulfills one or more of the following criteria: substances that are known to be, or may reasonably be anticipated to be, carcinogenic, mutagenic, teratogenic, or neurotoxic, causes reproductive dysfunction, is acutely or chronically toxic, or causes the threat of adverse environmental effects through ambient concentrations, bioaccumulation, or atmospheric deposition. lndene is acutely toxic with effects on the upper respiratory system (mucous membranes), pulmonary irritation, liver and kidney effects. Molecular Weight (g/mol): 116.15 Synonyms: Indonaphthene U.S. EPA Carcinogenic Classification (IRIS): Not listed on IRIS. PBT: Not listed as persistent, bioaccumulative and toxic. NTP: Not listed by the National Toxicology Program. HAP: Not listed as a hazardous air pollutant (HAP) by U.S. EPA. 112r: Not listed under Section 112(r) of the Clean Air Act. ACGIH: TLV: 10 ppm or 47,505 µg/m3. Critical effects include upper respiratory irritation or damage, pulmonary irritation, and liver and kidney effects. HSDB: Listed in the Hazardous Substances Data Bank. Inhalation of indene vapors is expected to cause irritation of mucous membranes. International IARC: Not listed by International Agency for Research on Cancer (IARC). ATSDR (MRL): Not listed by ATSDR. DataSheet Indene.wpd 2/2 Reference Material 1. American Conference of Governmental Industrial Hygienists (ACGIH) 2006. TLVs and BEIs: -

Using SMILES Strings for the Description of Chemical Connectivity in the Crystallography Open Database
Quirós et al. J Cheminform (2018) 10:23 https://doi.org/10.1186/s13321-018-0279-6 RESEARCH ARTICLE Open Access Using SMILES strings for the description of chemical connectivity in the Crystallography Open Database Miguel Quirós1* , Saulius Gražulis2,3 , Saulė Girdzijauskaitė3, Andrius Merkys2 and Antanas Vaitkus2 Abstract Computer descriptions of chemical molecular connectivity are necessary for searching chemical databases and for predicting chemical properties from molecular structure. In this article, the ongoing work to describe the chemical connectivity of entries contained in the Crystallography Open Database (COD) in SMILES format is reported. This col- lection of SMILES is publicly available for chemical (substructure) search or for any other purpose on an open-access basis, as is the COD itself. The conventions that have been followed for the representation of compounds that do not ft into the valence bond theory are outlined for the most frequently found cases. The procedure for getting the SMILES out of the CIF fles starts with checking whether the atoms in the asymmetric unit are a chemically accept- able image of the compound. When they are not (molecule in a symmetry element, disorder, polymeric species,etc.), the previously published cif_molecule program is used to get such image in many cases. The program package Open Babel is then applied to get SMILES strings from the CIF fles (either those directly taken from the COD or those produced by cif_molecule when applicable). The results are then checked and/or fxed by a human editor, in a computer-aided task that at present still consumes a great deal of human time. -
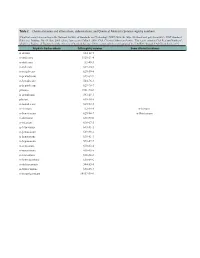
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Carcinogens CAS DOT SHHC Sources Number Chemical Name
2010 Right to Know Special Health Hazardous Substance List Substance Common Name Carcinogens CAS DOT SHHC Sources Number Chemical Name 3140 # ACEPHATE 30560-19-1 2783 CA 3 6 8 17 18 PHOSPHORAMIDOTHIOIC ACID, ACETYL-, O,S-DIMETHYL ESTER 0001 # ACETALDEHYDE 75-07-0 1089 CA MU TE F4 1 2 3 4 5 6 7 8 R2 15 17 18 20 21 ACETALDEHYDE 22 2890 # ACETAMIDE 60-35-5 3077 CA 3 6 7 17 18 20 ACETAMIDE 0010 # 2-ACETYLAMINOFLUORENE 53-96-3 CA MU 1 4 5 6 18 20 21 ACETAMIDE, N-9H-FLUOREN-2-YL- 0022 # ACRYLAMIDE 79-06-1 2074 CA R2 1 2 3 4 5 6 7 8 15 17 18 19 20 2-PROPENAMIDE 21 0024 # ACRYLONITRILE 107-13-1 1093 CA TE F3 R2 1 2 3 4 5 6 7 8 14 15 17 18 19 2-PROPENENITRILE 20 21 22 3142 # AF- 2 3688-53-7 CA 7 2-FURANACETAMIDE, .alpha.-[(5-NITRO-2-FURANYL)METHYLENE]- 0029 # AFLATOXINS 1402-68-2 CA MU TE 5 7 AFLATOXINS 0033 # ALDRIN 309-00-2 2761 CA TE 1 2 3 4 6 7 8 14 17 18 19 20 21 1,4:5,8-DIMETHANONAPHTHALENE, 1,2,3,4,10,10-HEXACHLORO-1,4,4a,5,8,8aHEXAHYDRO(1R,4S,4aS,5S,8R,8aR)-rel- Page 1 of 55 2010 Right to Know Special Health Hazardous Substance List Substance Common Name Carcinogens CAS DOT SHHC Sources Number Chemical Name 0039 # ALLYL CHLORIDE 107-05-1 1100 CA F3 1 2 3 4 6 7 8 15 17 18 20 1-PROPENE, 3-CHLORO- 0069 # 2-AMINOANTHRAQUINONE 117-79-3 CA MU 5 6 7 18 9,10-ANTHRACENEDIONE, 2-AMINO- 4012 # 1-AMINO-2,4-DIBROMOANTHRAQUINONE 81-49-2 CA 5 9,10-ANTHRACENEDIONE, 1-AMINO-2,4-DIBROMO- 0072 # 4-AMINODIPHENYL 92-67-1 CA MU 1 2 4 5 6 7 18 20 [1,1'-BIPHENYL]-4-AMINE 0076 # 1-AMINO-2-METHYLANTHRAQUINONE 82-28-0 CA 5 6 7 18 9,10-ANTHRACENEDIONE, 1-AMINO-2-METHYL- -

Furans in the Reactions with Electrophiles
Theoretical Study of Benzofused Thieno[3,2-b]furans in the reactions with electrophiles Ausra Vektariene1*, Gytis Vektaris1, Jiri Svoboda2 (1) Institute of Theoretical Physics and Astronomy of Vilnius University, A. Gostauto 12, LT- 01108 Vilnius, Lithuania (2) Department of Organic Chemistry, Prague Institute of Chemical Technology, Technicka 5, CZ-166 28 Prague 6, Czech Republic E-mail: [email protected] Keywords: Benzofused thieno[3,2-b]furans, electrophile, theoretical study, DFT. Abstract Calculations of traditional HF and DFT based reactivity descriptors are reported for the benzofused thieno[3,2-b]furans in order to get insight into the factors determining the exact nature of its interactions with electrophiles. Global descriptors such as chemical potential, molecular hardness, electrophilicity, frontier molecular orbital energies and shapes, the condensed Fukui functions were determined and used to identify the differences in the stability and reactivity of heterocycles. In general calculated values lead to the conclusion that heterocyclic system in thieno[3,2-b]benzofuran is more aromatic and stable than in isomeric benzothieno[3,2-b]furan. Theoretical results are in complete agreement with the experimental results showing exceptional reactivity of C(2) atom for both isomers. The bond order uniformity analysis, local ionization energy and electrostatic potential energy maps revealed structural and reactivity differences of isomeric thieno[3,2-b]furans. Benzothieno[3,2-b]furan structurally could be analogues with molecule of aromatic benzothiophene substituted with C(2)-C(3) vinylic moiety. Contrary, calculated values for the of thieno[3,2-b]benzofuran shows delocalized π–electron surface that reports stable aromatic system between benzene ring and thiene heterocycle. -

Coal Tar Creosote
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization. Concise International Chemical Assessment Document 62 COAL TAR CREOSOTE Please note that the layout and pagination of this pdf file are not identical to the document being printed First draft prepared by Drs Christine Melber, Janet Kielhorn, and Inge Mangelsdorf, Fraunhofer Institute of Toxicology and Experimental Medicine, Hanover, Germany Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 2004 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. -

Haloselectivity of Heterocycles Will Gutekunst
Baran Group Meeting Haloselectivity of Heterocycles Will Gutekunst Background SN(ANRORC) Addition of Nuclophile, Ring Opening, Ring Closure Polysubstituted heterocycles represent some of the most important compounds in the realm of pharmaceutical and material sciences. New and more efficient ways to selectively produce these Br Br molecules are of great importance and one approach is though the use of polyhalo heterocycles. Br NaNH2 N N Consider: Ar N 3 Ar2 NH3(l), 90% NH2 3 Halogenations H NH2 H N CO2Me Ar H 3 Suzuki Couplings 1 N CO2Me - Br H Ar 3 Ar2 1 Triple Halogenation NH2 NH CO Me N N 2 Ar1 H 1 Triple Suzuki Coupling N CO2Me N NH H NH2 Ar H 3 Ar2 1 Triple C-H activation? CO Me N 2 Ar1 H N CO2Me Cross Coupling H Virtually all types of cross coupling have been utilized in regioselective cross coupling reactions: Nucleophilic Substitution Kumada, Negishi, Sonogashira, Stille, Suzuki, Hiyama, etc. SNAr or SN(AE) In all of these examples, the oxidative addition of the metal to the heterocycle is the selectivity determining steps and is frequently considered to be irreversible. This addition highly resembles a nucleophilic substitution and it frequently follows similar regioselectivities in traditional S Ar reactions. Nu N The regioselectivity of cross coupling reaction in polyhalo heterocycles do not always follow the BDE's Nu of the corresponding C-X bonds. N X N X N Nu 2nd 2nd Meisenheimer Complex 1st Br Br 88.9 83.2 S (EA) Br 88.9 N 87.3 Br O O via: st OMe 1 Br NaNH , t-BuONa 2 N N pyrrolidine + Merlic and Houk have determined that the oxidative addition in palladium catalyzed cross coupling N N THF, 40ºC N reactions is determined by the distortion energy of the C-X bond (related to BDE) and the interaction of N the LUMO of the heterocycle to the HOMO of the Pd species. -

Modeling the Hydrodesulfurization Reaction at Nickel. Unusual Reactivity of Dibenzothiophenes Relative to Thiophene and Benzothiophene
7606 J. Am. Chem. Soc. 1999, 121, 7606-7617 Modeling the Hydrodesulfurization Reaction at Nickel. Unusual Reactivity of Dibenzothiophenes Relative to Thiophene and Benzothiophene David A. Vicic and William D. Jones* Contribution from the Department of Chemistry, The UniVersity of Rochester, Rochester, New York 14627-0216 ReceiVed February 25, 1999 Abstract: The nickel hydride dimer [(dippe)NiH]2 (1) was found to react with a variety of organosulfur substrates under mild conditions leading to C-S bond insertion adducts. The transition-metal insertions into the C-S bonds of thiophene, benzothiophene, and dibenzothiophene are all reversible, and lead to new organometallic complexes when dissolved in hydrocarbon solvent. (dippe)Ni(η2-C,S-dibenzothiophene) (6) converts to four new organometallic species in a unique desulfurization reaction that is believed to proceed via the intermediacy of the late-metal terminal sulfido complex (dippe)NidS(23). Independent synthetic routes to the desulfurization products have also provided an entry into the preparation of a variety of nickel-sulfur complexes such as (dippe)Ni(SH)2, (dippe)2Ni2(µ-H)(µ-S), [(dippe)2Ni2(µ-H)(µ-S)][PF6], and [(dippe)Ni(µ-S)]2, all of which have been structurally characterized. The reactivity of 1 with 4-methyldibenzothiophene, 4,6-dimethyldiben- zothiophene, 1,9-dimethyldibenzothiophene, thioxanthene, and thianthrene is also presented. Introduction desulfurize the more stubborn organosulfur compounds such as dibenzothiophene upon acidic workup5 and in the presence Raney nickel has seen widespread use in the desulfurization of reducing agents.6 These results, along with recent successes of organic and organometallic compounds.1 Catalytic desulfu- in platinum HDS modeling studies,7 encouraged us to prepare rization with Raney nickel under mild laboratory conditions is a new nickel hydride complex and examine the reactivity of not feasible, however, because of the chemical inertness of the this reagent toward carbon-sulfur bond activation in thiophenic resulting nickel sulfide that is produced. -

Chlordane and Heptachlor
CHLORDANE AND HEPTACHLOR Chlordane and heptachlor were considered together because of their close structural similarity and because technical-grade products each contain about 10–20% of the other compound. These substances were considered by previous working groups, in 1978 (IARC, 1979), 1987 (IARC, 1987) and 1990 (IARC, 1991). Since that time, new data have become available, and these have been incorporated into the monograph and taken into consideration in the present evaluation. 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Synonyms, structural and molecular data Chemical Abstract Services Registry numbers, names and synonyms of chlordane and heptachlor and its epoxide are given in Table 1. Cl Cl Cl Cl Cl Cl Cl Cl C10H6Cl8 Chlordane Relative molecular mass: 409.8 Cl Cl Cl Cl Cl Cl Cl C10H5Cl7 Heptachlor Relative molecular mass: 373.5 –411– 412 IARC MONOGRAPHS VOLUME 79 Table 1. Chemical Abstract Services Registry numbers, names and synonyms of chlordane, heptachlor and its epoxide Name CAS Reg. Nosa Chem. Abstr. namesb and synonyms Chlordane 57-74-9 ENT 9932; 1,2,4,5,6,7,8,8-octachloro-2,3,3a,4,7,7a- (39400-80-1); hexahydro-4,7-methano-1H-indene; 1,2,4,5,6,7,8,8- 53637-13-1) octachloro-2,3,3a,4,7,7a-hexahydro-4,7-methano- indene (IUPAC); octachloro-4,7-methanotetrahydro- indane; 1,2,4,5,6,7,8,8-octachloro-3a,4,7,7a-tetra- hydro-4,7-methanoindan; OMS 1437 Technical-grade 12789-03-6 chlordane cis-Chlordane 5103-71-9 α-Chlordan; α-chlordane; cis-chlordan; (152322-29-7; (1α,2α,3aα,4β,7β,7aα)-1,2,4,5,6,7,8,8-octachloro- 22212-52-8;