Introduction of Fluorine and Fluorine- Containing Functional Groups
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
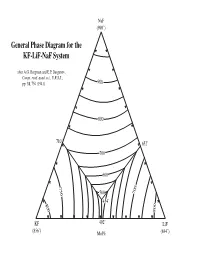
General Phase Diagram for the KF-Lif-Naf System
NaF (990˚) General Phase Diagram for the KF-LiF-NaF System after A.G. Bergman and E.P. Dergunov, Compt. rend. acad. sci., U.R.S.S., pp. 31, 754 (1941). 900 800 710˚ 652˚ 700 600 750 500 700 454˚ 800 800 KF 492˚ LiF (856˚) Mol% (844˚) Sodium Fluoride NaF 950˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 940˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 930˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 920˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 910˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 900˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 890˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 880˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 870˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 860˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 850˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 840˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 830˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 820˚ KF LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 810˚ 810˚ ˚ KF 810 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 800˚ 800˚ ˚ KF 800 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 790˚ 790˚ ˚ KF 790 LiF Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 780˚ 780˚ ˚ LiF KF 780 Potassium Fluoride Lithium Fluoride Sodium Fluoride NaF 770˚ 770˚ ˚ LiF KF 770 Potassium Fluoride -

Acidity, Basicity, and Pka 8 Connections
Acidity, basicity, and pKa 8 Connections Building on: Arriving at: Looking forward to: • Conjugation and molecular stability • Why some molecules are acidic and • Acid and base catalysis in carbonyl ch7 others basic reactions ch12 & ch14 • Curly arrows represent delocalization • Why some acids are strong and others • The role of catalysts in organic and mechanisms ch5 weak mechanisms ch13 • How orbitals overlap to form • Why some bases are strong and others • Making reactions selective using conjugated systems ch4 weak acids and bases ch24 • Estimating acidity and basicity using pH and pKa • Structure and equilibria in proton- transfer reactions • Which protons in more complex molecules are more acidic • Which lone pairs in more complex molecules are more basic • Quantitative acid/base ideas affecting reactions and solubility • Effects of quantitative acid/base ideas on medicine design Note from the authors to all readers This chapter contains physical data and mathematical material that some readers may find daunting. Organic chemistry students come from many different backgrounds since organic chemistry occu- pies a middle ground between the physical and the biological sciences. We hope that those from a more physical background will enjoy the material as it is. If you are one of those, you should work your way through the entire chapter. If you come from a more biological background, especially if you have done little maths at school, you may lose the essence of the chapter in a struggle to under- stand the equations. We have therefore picked out the more mathematical parts in boxes and you should abandon these parts if you find them too alien. -

The Synthesis of Molecules Containing Quaternary Stereogenic Centers Via the Intramolecular Asymmetric Heck Reaction
The Synthesis of Molecules Containing Quaternary Stereogenic Centers via the Intramolecular Asymmetric Heck Reaction Reported by Eric P. Gillis April 19, 2007 INTRODUCTION The enantioselective synthesis of compounds containing quaternary stereocenters continues to be a challenging endeavor and is a field of intense interest and development.1,,,,2 3 4 5 From the perspective of methods development, the asymmetric synthesis of compounds containing quaternary stereocenters poses a distinct challenge due to the sterically congested environment in which C-C bonds must be formed and stereochemical information must be transferred. Existing asymmetric methods have been applied successfully in desymmetrization processes that transform prochiral quaternary centers into stereodefined quaternary centers. However, for processes in which a quaternary stereocenter is created directly via a C-C bond forming reaction, only a few asymmetric reactions have been fruitful. The diversity in the array of methods that exist for the synthesis of compounds containing quaternary stereocenters is necessitated in part because of the number of distinct quaternary stereocenter architectures that are possible (Scheme 1). The construction of acyclic quaternary stereocenters is particularly challenging due to the number of degrees of freedom associated with these structures. However, promising results for the construction of these stereocenters via allylation,6 allylic alklyation,7 and Michael addition8 have been reported recently. For the synthesis of cyclic quaternary stereocenters a number of methods have been employed successfully including the intramolecular Heck reaction,9 alkylation,10 arylation,11 and Michael addition12. Fused bicyclic quaternary stereocenters are generally accessed via the desymmetrization of a prochiral quaternary center. In this regard, theoretically any SCHEME 1. -

Vibrationally Excited Hydrogen Halides : a Bibliography On
VI NBS SPECIAL PUBLICATION 392 J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards National Bureau of Standards Bldg. Library, _ E-01 Admin. OCT 1 1981 191023 / oO Vibrationally Excited Hydrogen Halides: A Bibliography on Chemical Kinetics of Chemiexcitation and Energy Transfer Processes (1958 through 1973) QC 100 • 1X57 no. 2te c l !14 c '- — | NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences" — Applied Radiation 2 — Quantum Electronics 1 — Electromagnetics 3 — Time 3 1 1 and Frequency — Laboratory Astrophysics — Cryogenics . -

Selective Syntheses of Difluoromethylene Compounds Via Difluorocarbene Catalyses
Selective Syntheses of Difluoromethylene Compounds via Difluorocarbene Catalyses 著者 青野 竜也 内容記述 この博士論文は全文公表に適さないやむを得ない事 由があり要約のみを公表していましたが、解消した ため、2017年10月2日に全文を公表しました。 year 2016 その他のタイトル ジフルオロカルベンを用いた触媒反応によるジフル オロメチレン化合物の選択的合成 学位授与大学 筑波大学 (University of Tsukuba) 学位授与年度 2015 報告番号 12102甲第7642号 URL http://hdl.handle.net/2241/00144162 Selective Syntheses of Difluoromethylene Compounds via Difluorocarbene Catalyses Tatsuya Aono February 2016 Selective Syntheses of Difluoromethylene Compounds via Difluorocarbene Catalyses Tatsuya Aono Doctoral Program in Chemistry Submitted to the Graduate School of Pure and Applied Sciences in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Science at the University of Tsukuba Contents Chapter 1 General Introduction 1 Chapter 2 O-Selective Difluoromethylation of Amides with Free Difluorocarbene 20 2.1. Introduction 21 2.2. Synthesis of Difluoromethyl Imidates 24 2.3. Mechanistic Considerations on O-Selective Difluoromethylation of Amides 31 2.4. Conclusion 33 2.5. Experimental Section 34 2.6. Reference 38 Chapter 3 Regioselective Syntheses of gem-Difluorocyclopentanone Derivatives with Transition Metal Difluorocarbene Complexes 39 3.1. Introduction 40 3.2. Domino Difluorocyclopropanation/Ring Expansion with Nickel Difluorocarbene Complex 45 3.3. [4 + 1] Cycloaddition with Copper Difluorocarbene Complex 58 3.4. Conclusion 66 3.5. Experimental Section 67 3.6. Reference 101 Chapter 4 Conclusion 104 List of Publications 105 Acknowledgement 106 Chapter 1 1. General Introduction Organofluorine compounds often exhibit unique properties and behaviors in comparison with nonfluorinated parent compounds, playing important roles as pharmaceuticals and agrochemicals. Because of the high bond dissociation energy of C–F bonds, organofluorine compounds are resistant to heat and chemicals, and stable to metabolism. In addition, organofluorine compounds have high lipophilicity. -

Trifluoromethane)
SAFETY DATA SHEET Halocarbon R-23 (Trifluoromethane) Section 1. Identification GHS product identifier : Halocarbon R-23 (Trifluoromethane) Chemical name : trifluoromethane Other means of : Fluoroform; Arcton 1; Fluoryl; Freon F-23; Freon 23; Genetron 23; Methyl trifluoride; R identification 23; Trifluoromethane; CHF3; Arcton; Halocarbon 23; UN 1984; Carbon trifluoride; Genetron HFC23; Propellant 23; Refrigerant 23 Product type : Liquefied gas Product use : Synthetic/Analytical chemistry. Synonym : Fluoroform; Arcton 1; Fluoryl; Freon F-23; Freon 23; Genetron 23; Methyl trifluoride; R 23; Trifluoromethane; CHF3; Arcton; Halocarbon 23; UN 1984; Carbon trifluoride; Genetron HFC23; Propellant 23; Refrigerant 23 SDS # : 001078 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Liquefied gas substance or mixture GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May cause frostbite. May displace oxygen and cause rapid suffocation. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -
![Triazene (H2NNNH) Or Triimide (HNHNNH) Markofçrstel,[A, D] Yetsedaw A](https://docslib.b-cdn.net/cover/4724/triazene-h2nnnh-or-triimide-hnhnnh-markof%C3%A7rstel-a-d-yetsedaw-a-184724.webp)
Triazene (H2NNNH) Or Triimide (HNHNNH) Markofçrstel,[A, D] Yetsedaw A
DOI:10.1002/cphc.201600414 Articles On the Formation of N3H3 Isomers in Irradiated Ammonia Bearing Ices:Triazene (H2NNNH) or Triimide (HNHNNH) MarkoFçrstel,[a, d] Yetsedaw A. Tsegaw,[b] Pavlo Maksyutenko,[a, d] Alexander M. Mebel,[c] Wolfram Sander,[b] and Ralf I. Kaiser*[a, d] The remarkable versatility of triazenesinsynthesis, polymer theoretical studies with our novel detection scheme of photo- chemistry and pharmacology has led to numerousexperimen- ionization-driven reflectron time-of-flight mass spectroscopy tal and theoretical studies.Surprisingly,only very little is we can obtain information on the isomersoftriazene formed known aboutthe most fundamental triazene:the parentmole- in the films. Using isotopically labeled starting material, we can cule with the chemical formula N3H3.Here we observe molecu- additionally gain insightinthe formation pathways of the iso- lar,isolated N3H3 in the gas phase after it sublimes from ener- mers of N3H3 under investigation and identify the isomers getically processed ammonia and nitrogen films. Combining formedastriazene (H2NNNH) andpossibly triimide(HNHNNH). 1. Introduction During the last decades, triazenes—a class of organic mole- life time of at least 1mswas also inferred as an intermediate cules carrying the =N N=N moiety—have received substan- in the radiolysis of an aqueous solution of hydrazine based on À À tial attention both from the theoretical and organic chemistry asingle absorption feature at 230 nm.[6] The cyclic isomer of [1] communities. Derived from cis-and trans-triazene (HN=NNH2 ; triazene, cyclotriazane, was first reported crystallographically in Scheme1), the substituted counterparts have significant appli- zeolite A, where it was stabilized by asilver cation as [1a,c] [1d] + [7] + cations in synthetic chemistry, polymer science, and phar- Ag(N3H3) . -

UNITED STATES PATENT of FICE 2,640,086 PROCESS for SEPARATING HYDROGEN FLUORIDE from CHLORODFLUORO METHANE Robert H
Patented May 26, 1953 2,640,086 UNITED STATES PATENT of FICE 2,640,086 PROCESS FOR SEPARATING HYDROGEN FLUORIDE FROM CHLORODFLUORO METHANE Robert H. Baldwin, Chadds Ford, Pa., assignor to E. H. du Pont de Nemours and Company, Wi inington, Del, a corporation of Delaware No Drawing. Application December 15, 1951, Serial No. 261,929 9 Claims. (C. 260-653) 2 This invention relates to a process for Sep These objects are accomplished essentially by arating hydrogen fluoride from monochlorodi Subjecting a mixture of hydrogen fluoride and fluoronethane, and more particularly, separat Inonochlorodifluoromethane in the liquid phase ing these components from the reaction mixture to temperatures below 0° C., preferably at about obtained in the fluorination of chloroform with -30° C. to -50° C., at either atmospheric or hydrogen fluoride, Super-atmospheric pressures, together with from In the fluorination of chloroform in the prest about 0.25 mol to about 2.5 mols of chloroform ence Of a Catalyst, a reaction mixture is pro per mol of chlorodifluoronethane contained in duced which consists essentially of HCl, HF, the mixture and separating an upper layer rich CHCIF2, CHCl2F, CHCls, and CHF3. A method O in HF from a lower organic layer. The proceSS of Separating these components is disclosed in is operative with mixtures containing up to 77% U. S. Patent No. 2,450,414 which involves sep by weight of HF. arating the components by a special fractional It has been found that chloroform is substan distillation under appropriate temperatures and tially immiscible With EIF at temperatures be pressures. -

Ionic Compound Ratios Time: 1 -2 Class Periods
Collisions Lesson Plan Ionic Compound Ratios Time: 1 -2 class periods Lesson Description In this lesson, students will use Collisions to explore the formation of ionic compounds and compound ratios. Key Essential Questions 1. What makes up an ionic compound? 2. Are ionic compounds found in common ratios? Learning Outcomes Students will be able to determine the ionic compound ratio of an ionic compound. Prior Student Knowledge Expected Cations are postiviely charged ions and anions are negatively charged ions. Lesson Materials • Individual student access to Collisions on tablet, Chromebook, or computer. • Projector / display of teacher screen • Accompanying student resources (attached) Standards Alignment NGSS Alignment Science & Enginnering Practices Disciplinary Core Ideas Crosscutting Concepts • Developing and using • HS-PS-12. Construct and • Structure and Function models revise an explanation for the • Construcing explanations outcome of a simple chemical and designing solutions rection based on the outermost electron states of atoms, trends int he periodic table, and knowl- edge of the partterns of chemi- cal properties. www.playmadagames.com ©2018 PlayMada Games LLC. All rights reserved. 1 PART 1: Explore (15 minutes) Summary This is an inquiry-driven activity where students will complete the first few levels of the Collisions Ionic Bonding game to become introduced to the concept of ionic bonding and compound ratios. Activity 1. Direct students to log into Collisions with their individual username and password. 2. Students should enter the Ionic Bonding game and play Levels 1-6 levels. 3. Have your students answer the following questions during gameplay: 1. What combination of ions did you use to successfully match a target? 2. -

From Celebrex to a Novel Class of Phosphoinositide-Dependent Kinase 1 (Pdk-1) Inhibitors for Androgen-Independent Prostate Caner
FROM CELEBREX TO A NOVEL CLASS OF PHOSPHOINOSITIDE-DEPENDENT KINASE 1 (PDK-1) INHIBITORS FOR ANDROGEN-INDEPENDENT PROSTATE CANER DISSERTATION Presented in Partial Fulfillment of the requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Jiuxiang Zhu, M.S. * * * * * The Ohio State University 2005 Dissertation Committee: Approved by Professor Ching-Shih Chen, Advisor Professor Robert Brueggemeier Advisor Professor Pui-Kai (Tom) Li Graduate Program in Pharmacy Professor Matthew D. Ringel ABSTRACT Celebrex, a nonsteroidal anti-inflammatory drug (NSAID, cyclooxygenase-2 inhibitor), was reported to induce apoptosis in the prostate cancer cell line PC-3 at 50µM. Early research from our laboratory demonstrated that this apoptotic inducing effect was independent of its COX-2 inhibitory activity. Further investigation showed that PDK-1 was a major protein targeted by celebrex in PC-3 cells to induce apoptosis. However, celebrex was very weak in inhibiting PDK-1 with IC50 of 48µM. To improve its potency, two series of analogs were designed and synthesized. In the first series of 24 compounds, the 5-position methylphenyl moiety of celebrex was replaced by various aromatic ring systems to explore the optimal hydrophobic group. OSU02067 (IC50=9µM) with phenanthrene at the 5-position was the best inhibitor among this series and was selected as the lead compound for further modification. Enzyme kinetics study of PDK-1 inhibition by celebrex indicated that it competed with ATP for binding. Docking of OSU02067 into the ATP binding domain of PDK-1 showed that the sulfonamide moiety hydrogen bonded to hinge region residue Ala162. -

Studies on New Vasodilators, Ws-1228 a and B Ii. Structure and Synthesis
VOL. XXXV NO. 2 THE JOURNAL OF ANTIBIOTICS 157 STUDIES ON NEW VASODILATORS, WS-1228 A AND B II. STRUCTURE AND SYNTHESIS HIROKAZU TANAKA, KEIZO YOSHIDA, YOSHIKUNI ITOH and HIROSHI IMANAKA Fermentation Research Laboratories, Fujisawa Pharmaceutical Co., Ltd., Osaka, Japan (Received for publication October 26, 1981) The structure of new hypotensive vasodilators, WS-1228 A and B, produced by Strepto- myces aureofaciens, were determined as I and 2, respectively, on the basis of their spectral and chemical evidences. WS-1228 A (1), having N-hydroxytriazene moiety, was synthesized from (E,E,E)-2,4,7- undecatrienal (4) by condensation with hydrazine hydrate followed by nitrosation. In the course of screening for new biologically active compounds, we found that Streptomyces aureofaciens produces hypotensive vasodilators designated as WS-1228 A (1) and B (2). Taxonomy, isolation and characterization of these compounds have been reported in the preceding papery. This report describes the structure elucidations of WS-1228 A (1) and B (2) and the synthesis of 1. WS-1228 A (1) WS-1228 B (2) WS-1228 A (1) was isolated as yellow needles [mp 100 - 102°C (dec.)] which showed a positive color reaction to ferric chloride reagent. Elemental analysis and mass spectrum established the molecular formula of 1 as C11H17N3O. Absorption bands at 1612 and 1580 cm-1 in its IR spectrum (Fig. 1) and at 300 nm in its UV spectrum suggested the presence of the triene oxime moiety. The PMR spectrum (Fig. Fig. 1. IR spectrum of WS-1228 A (1) (Nujol). 158 THE JOURNAL OF ANTIBIOTICS FEB.