Amino Acids and Peptides I
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
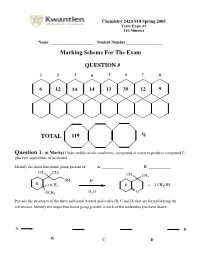
Marking Scheme for the Exam
Chemistry 2420 S10 Spring 2005 Term Exam #1 110 Minutes Name: ______________________ Student Number___________________ Marking Scheme For The Exam QUESTION # 1 2 3 4 5 6 7 8 6 12 14 14 13 39 12 9 TOTAL 119 % Question 1. (6 Marks) Under mildly acidic conditions, compound A reacts to produce compound E plus two equivalents of methanol. Identify the main functional group present in: A: ___________ E: ___________ CH3 CH3 CH 3 CH3 + NH2 H A OCH3 E + 2 CH3OH H O OCH3 2 N Provide the structures of the three additional neutral molecules (B, C and D) that are formed during the conversion. Identify the major functional group present in each of the molecules you have drawn. A E B C D Question 2. (12 Marks) Circle the structure that will best satisfy the given information. - the compound able to undergo an intramolecular cyclization to form a hemiacetal H H HO H HO OH O O O - the compound able to undergo an intramolecular cyclization to form an enamine H H H Me2N MeHN H2N O O O + - the produc t formed from treatment of 2-butanone with NaBD4 followed by a H3O work-up DO H HO D DO D HO H - the compound that would most likely be coloured O H O O - the compound capable of undergoing mutarotation O O O OH OCH3 OH CH3 H CH 3 - the compound that would not react with MeMgBr O O O OH OCH3 OH CH3 H CH3 - the compound bes t described as “anti-aromatic”: - the most stable carbocation + + + Cl Cl Cl OCH 3 H NO2 H H H Question 3. -

REFERENCE ONLY UNIVERSITY of LONDON THESIS This Thesis
REFERENCE ONLY UNIVERSITY OF LONDON THESIS Degree Year Name of Author 0" * COPYRIGHT This is a thesis accepted for a Higher Degree of the University of London. It is an unpublished typescript and the copyright is held by the author. All persons consulting the thesis must read and abide by the Copyright Declaration below. COPYRIGHT DECLARATION I recognise that the copyright of the above-described thesis rests with the author and that no quotation from it or information derived from it may be published without the prior written consent of the author. LOANS Theses may not be lent to individuals, but the Senate House Library may lend a copy to approved libraries within the United Kingdom, for consultation solely on the premises of those libraries. Application should be made to: Inter-Library Loans, Senate House Library, Senate House, Malet Street, London WC1E 7HU. REPRODUCTION University of London theses may not be reproduced without explicit written permission from the Senate House Library. Enquiries should be addressed to the Theses Section of the Library. Regulations concerning reproduction vary according to the date of acceptance of the thesis and are listed below as guidelines. A. Before 1962. Permission granted only upon the prior written consent of the author. (The Senate House Library will provide addresses where possible). B. 1962- 1974. In many cases the author has agreed to permit copying upon completion of a Copyright Declaration. C. 1975 - 1988. Most theses may be copied upon completion of a Copyright Declaration. D. 1989 onwards. Most theses may be copied. This thesis comes within category D. -

Part 1 in Our Series of Carbohydrate Lectures. in This Section, You Will Learn About Monosaccharide Structure
Welcome to Part 1 in our series of Carbohydrate lectures. In this section, you will learn about monosaccharide structure. The building blocks of larger carbohydrate polymers. 1 First, let’s review why learning about carbohydrates is important. Carbohydrates are used by biological systems as fuels and energy resources. Carbohydrates typically provide quick energy and are one of the primary energy storage forms in animals. Carbohydrates also provide the precursors to other major macromolecules within the body, including the deoxyribose and ribose required for nucleic acid biosynthesis. Carbohydrates can also provide structural support and cushioning/shock absorption, as well as cell‐cell communication, identification, and signaling. 2 Carbohydrates, as their name implies, are water hydrates of carbon, and they all have the same basic core formula (CH2O)n and are always found in the ratio of 1 carbon to 2 hydrogens to 1 oxygen (1:2:1) making them easy to identify from their molecular formula. 3 Carbohydrates can be divided into subcategories based on their complexity. The simplest carbohydrates are the monosaccharides which are the simple sugars required for the biosynthesis of all the other carbohydrate types. Disaccharides consist of two monosaccharides that have been joined together by a covalent bond called the glycosidic bond. Oligosaccharides are polymers that consist of a few monosaccharides covalently linked together, and Polysaccharides are large polymers that contain hundreds to thousands of monosaccharide units all joined together by glycosidic bonds. The remainder of this lecture will focus on monosaccharides 4 Monosaccharides all have alcohol functional groups associated with them. In addition they also have one additional functional group, either an aldehyde or a ketone. -

Lipids, Carbohydrates, Nucleobases & DNA
Chemistry 2050 “Introduction to Organic Chemistry” Fall Semester 2011 Dr. Rainer Glaser Examination #5: The Final “Lipids, Carbohydrates, Nucleobases & DNA.” Monday, December 12, 2011, 10 am – 12 pm. Name: Answer Key Question 1. Lipids and Detergents. 20 Question 2. Glyceraldehyde and Carbohydrates. 20 Question 3. Nucleobases of DNA and RNA. 20 Question 4. Ribose and Deoxyribose and Their Phosphate Esters. 20 Question 5. Single and Double Strands of DNA. 20 Total 100 — 1 — Question 1. Lipids and Detergents. (20 points) (a) Draw the line segment drawing of glyceryl distearoleate. This fat is the _TRI-ester formed between one molecule of the triol __GLYCEROL_, two molecules of the fatty acid stearic acid, H3C−(CH2)16−COOH, and one molecule of the unsaturated fatty acid oleic acid, H3C−(CH2)7−CH=CH−(CH2)7−COOH. In your drawing, attach the oleic acid to one of the primary alcohols of the triol. Clearly indicate whether the alkene in oleic acid is cis or trans. To convert this fat into soap, one would cook the fat with aqueous NaOH solution, a process that is called __SAPONIFICATION__. (12 points) (b) A simple micelle is shown schematically on the right. Each circle signifies a _________ (polar, nonpolar) head-group, and each wiggly line signifies a ________ (polar, nonpolar) alkyl chain. In the case of “normal” soap, the head- group is a ___CARBOXYLATE__ anion. Indicate where we would find the associated cations (i.e., sodium cations; draw a few in the scheme). Water is on the __________ (inside, outside) of this micelle and fats will accumulate on the _________ (inside, outside) of this micelle. -

Organic Chemistry by Robert C
(2/94)(6,7,9/95)(8,9/97)(12/99)(1/00) Neuman Chapter 4 Chapter 4 Stereochemistry from Organic Chemistry by Robert C. Neuman, Jr. Professor of Chemistry, emeritus University of California, Riverside [email protected] <http://web.chem.ucsb.edu/~neuman/orgchembyneuman/> Chapter Outline of the Book ************************************************************************************** I. Foundations 1. Organic Molecules and Chemical Bonding 2. Alkanes and Cycloalkanes 3. Haloalkanes, Alcohols, Ethers, and Amines 4. Stereochemistry 5. Organic Spectrometry II. Reactions, Mechanisms, Multiple Bonds 6. Organic Reactions *(Not yet Posted) 7. Reactions of Haloalkanes, Alcohols, and Amines. Nucleophilic Substitution 8. Alkenes and Alkynes 9. Formation of Alkenes and Alkynes. Elimination Reactions 10. Alkenes and Alkynes. Addition Reactions 11. Free Radical Addition and Substitution Reactions III. Conjugation, Electronic Effects, Carbonyl Groups 12. Conjugated and Aromatic Molecules 13. Carbonyl Compounds. Ketones, Aldehydes, and Carboxylic Acids 14. Substituent Effects 15. Carbonyl Compounds. Esters, Amides, and Related Molecules IV. Carbonyl and Pericyclic Reactions and Mechanisms 16. Carbonyl Compounds. Addition and Substitution Reactions 17. Oxidation and Reduction Reactions 18. Reactions of Enolate Ions and Enols 19. Cyclization and Pericyclic Reactions *(Not yet Posted) V. Bioorganic Compounds 20. Carbohydrates 21. Lipids 22. Peptides, Proteins, and α−Amino Acids 23. Nucleic Acids ************************************************************************************** -
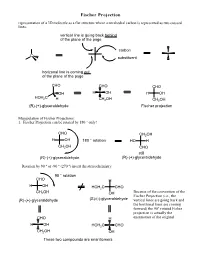
Fischer Projection Representation of a 3D Molecule As a Flat Structure Where a Tetrahedral Carbon Is Represented As Two Crossed Lines
Fischer Projection representation of a 3D molecule as a flat structure where a tetrahedral carbon is represented as two crossed lines. vertical line is going back behind of the plane of the page carbon C substituent horizonal line is coming out of the plane of the page CHO CHO CHO OH H OH H OH HOH2C H CH2OH CH2OH (R)-(+)-glyceraldehyde Fischer projection Manipulation of Fischer Projections: 1. Fischer Projection can be rotated by 180 ° only! CHO CH2OH H OH 180 ° rotation HO H CH2OH CHO still (R)-(+)-glyceraldehyde (R)-(+)-glyceraldehyde Rotation by 90 ° or -90 ° (270 °) invert the stereochemistry 90 ° rotation CHO H H OH HOH2C CHO CH2OH OH Because of the convention of the Fischer Projection (i.e., the (R)-(+)-glyceraldehyde (S)-(-)-glyceraldehyde vertical lines are going back and the horizonal lines are coming forward) the 90° rotated Fisher projection is actually the CHO H enantiomer of the original H OH HOH2C CHO CH2OH OH These two compounds are enantiomers 2. If one group of the Fischer projection is held steady, the other groups can be rotated either clockwise or counter clockwise. hold this group steady CHO CHO H OH HOH2C H CH2OH OH still (R)-(+)-glyceraldehyde (R)-(+)-glyceraldehyde Assigning R and S-configurations to Fischer projections 1. Assign priorities to the four substituents according to the Cahn-Ingold-Prelog convention. 2. Perform the two allowed manipulations of the Fischer projection to place the lowest priority group on one of the vertical positions (either top or bottom) 3. If the priorities of the other three groups (1-2-3) proceed clockwise, the stereogenic center is assigned as R. -

Hermann Emil Fischer: Life and Achievements
GENERAL ARTICLE Hermann Emil Fischer: Life and Achievements G Nagendrappa Emil Fischer, considered as one of the greatest chemists of all times, carried out much of the fundamental work on purines, sugars, proteins, stereochemistry and several other areas of chemistry during the late nineteenth and early twentieth century. Because most of these are biological molecules, he is known as the ‘Father of Biochemistry’. His achievements in G Nagendrappa was a Professor of Organic chemical synthesis and analytical skills were much ahead of Chemistry at Bangalore his times. He was the second to get the Nobel Prize in Chem- University, and Head of istry in 1902. the Department of Medicinal Chemistry, Sri Introduction Ramachandra (Medical) University, Chennai. He is Carbohydrates, proteins, fats, and nucleic acids are the four major currently in Jain Univer- sity, Bangalore. He chemical constituents of living organisms. These four classes of continues to teach and do organic compounds are the main players in the emergence and research. His work is in existence of life. Extensive pioneering contributions to the devel- the area of organosilicon opment of all these areas of organic chemistry and biochemistry chemistry, synthetic and mechanistic organic were made by Emil Fischer through his more than four decades of chemistry, and clay- brilliant research work starting from the early 1870s. His uncanny catalysed organic reactions skills in analytical and synthetic work, his extraordinary under- (Green Chemistry). standing of the enormous amount of experimental results and their correct interpretation laid a solid foundation for the chemis- try of these biologically important molecules. Though his work essentially constituted analytical and synthetic organic chemis- try, its reach extended to other areas, particularly stereochemis- try, biochemistry, physiology and medicine. -

Biochemical Regulation of Brassica Napus Diacylglycerol Acyltransferase 1
! Biochemical Regulation of Brassica napus Diacylglycerol Acyltransferase 1 by Kristian Mark P. Caldo A thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Plant Science Department of Agricultural, Food and Nutritional Science University of Alberta ©Kristian Mark P. Caldo, 2017 ! ! ABSTRACT Diacylglycerol acyltransferase 1 (DGAT1) is an integral membrane enzyme catalyzing the final and committed step in acyl-CoA-dependent triacylglycerol (TAG) biosynthesis. Previous metabolic control analysis showed that DGAT1 plays a substantial role in modulating the flow of carbon into seed oil in Brassica napus. In B. napus and many other organisms, DGAT1 has been used as a biotechnological tool to boost oil accumulation. Despite its key role in primary metabolism and biotechnological importance, no detailed structure-function information is available on the enzyme. This doctoral thesis aimed to shed light on the mechanism of action and regulation of DGAT1 by employing various techniques in molecular biology, and protein and lipid biochemistry. The first part of this dissertation involved the development of a purification protocol for recombinant B. napus DGAT1 (BnaDGAT1) produced in Saccharomyces cerevisiae. The optimized protocol included solubilization of membrane-bound BnaDGAT1 in n-dodecyl-!-D-maltopyranoside micelles (DDM), cobalt affinity chromatography, and size-exclusion chromatography. BnaDGAT1 was eluted mainly as a dimer from the last chromatographic step. The purified enzyme was well-folded, intact, and active and utilized acyl-CoAs which represent the major fatty acids in canola- type B. napus seed oil. In the second part of this dissertation, the hydrophilic N-terminal domain of BnaDGAT1 was identified as the enzyme’s regulatory domain that is not essential for catalysis. -
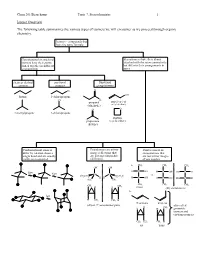
Isomer Overview the Following Table Summarizes the Various Types Of
Chem 201/Beauchamp Topic 7, Stereochemistry 1 Isomer Overview The following table summarizes the various types of isomers we will encounter as we proceed through organic chemistry. Isomers - compounds that have the same formula Constitutional or structural Stereoisomers have their atoms isomers have their atoms attached with the same connectivity, joined together in different but differ in their arrangements in arrangements space chain or skeletal positional functional isomers isomers group isomers Cl O OH butane 1-chloropropane Cl H propanal prop-2-en-1-ol (aldehyde) (allyl alcohol) O O 2-methylpropane 2-chloropropane oxatane propanone (cyclic ether) (ketone) Conformational isomers Enantiomers are mirror Diastereomers are differ by rotation about a image reflections that stereoisomers that single bond and are usually are not superimposable are not mirror images easily interconverted. (different). of one another. CH a. 3 CH3 CH3 OH OH fast H C OH H C OH HO C H fast C C vs. CH3CH2 H2CH3C H H H C OH H C OH HO C H H3C CH3 CH 3 CH3 CH3 CH3 CH H H 3 meso (dl) enantiomers b. fast H CH3 CH3 H E or trans Z or cis (dl) or () enantiomer pairs also called H CH H H 3 geometric isomers and cis/trans isomers CH3 CH3 CH3 H cis trans Chem 201/Beauchamp Topic 7, Stereochemistry 2 Essential Vocabulary of Stereochemistry – terms to know for this topic Chiral center – a tetrahedral atom with four different groups attached, a single chiral center is not superimposable on its mirror image, also called a stereogenic center, and asymmetric center Stereogenic center – any center where interchange of two groups produces stereoisomers (different structures), could be a chiral center producing R/S differences, or an alkene producing E/Z differences or cis/trans in a ring Absolute configuration (R and S) – the specific 3 dimensional configuration about a tetrahedral shape, determined by assigning priorities (1-4) based on the highest atomic number of an atom at the first point of difference. -

Examining the Relationship Between 2D Diagrammatic Conventions and Students' Success on Representational Translation Tasks In
Chemistry Education Research and Practice Examining the Relationship between 2D Diagrammatic Conventions and Students’ Success on Representational Translation Tasks in Organic Chemistry Journal: Chemistry Education Research and Practice Manuscript ID: RP-ART-08-2014-000169.R2 Article Type: Paper Date Submitted by the Author: 07-Nov-2014 Complete List of Authors: Olimpo, Jeffrey; University of Northern Colorado, School of Biological Sciences; University of Maryland, College Park, Teaching and Learning, Policy and Leadership Kumi, Bryna; University of Maryland, Chemistry & Biochemistry Wroblewski, Richard; University of Maryland, College Park, Chemistry & Biochemistry Dixon, Bonnie; University of Maryland, College Park, Chemistry & Biochemistry Page 1 of 21 Chemistry Education Research and Practice DIAGRAMMATIC CONVENTIONS IN ORGANIC CHEMISTRY 1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Examining the Relationship between 2D Diagrammatic Conventions and Students’ Success on 23 24 Representational Translation Tasks in Organic Chemistry 25 12 3 3 3* 26 Jeffrey T. Olimpo , Bryna C. Kumi , Richard Wroblewski , & Bonnie L. Dixon 27 1School of Biological Sciences; University of Northern Colorado, Greeley, CO 80639, USA 28 2Department of Teaching and Learning, Policy and Leadership; University of Maryland, College 29 Park, MD 20742, USA 30 3 31 Department of Chemistry and Biochemistry; University of Maryland, College Park, MD 20742, 32 USA 33 34 35 Jeffrey T. Olimpo: [email protected] 36 37 Bryna C. Kumi: [email protected] 38 Richard Wroblewski: [email protected] 39 40 *Corresponding Author: e-mail: [email protected] 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Chemistry Education Research and Practice Page 2 of 21 DIAGRAMMATIC CONVENTIONS IN ORGANIC CHEMISTRY 2 1 2 3 Abstract: Two-dimensional (2D) diagrams are essential in chemistry for conveying and communicating 4 key knowledge about disciplinary phenomena. -

Carbohydrate Nomenclature Worksheet KEY CHEM 8B, UCSC, Binder
Carbohydrate Nomenclature Worksheet KEY CHEM 8B, UCSC, Binder 1. Draw one example of each of the following types of monosaccharides (there may be several correct answers) and indicate the number of possible stereoisomers while keeping the same D/L configuration. (a) D-Aldotriose (b) L-Aldotriose (c) L-Ketotetrose CHO CHO CH2OH H OH HO H C O CH2OH CH2OH HO H 1 possible 1 possible CH2OH 1 possible (d) L-Aldopentose 4 possible CHO CHO CHO CHO H OH H OH HO H HO H H OH HO H H OH HO H HO H HO H HO H HO H CH2OH CH2OH CH2OH CH2OH (e) D-Ketohexose ANY ONE EXAMPLE IS CORRECT 4 possible FOR (d), (e), & (f). CH2OH I WOULDN'T EXPECT YOU TO CH2OH CH2OH CH2OH C O DRAW ALL STEREOISOMERS C O C O C O ON AN EXAM! H OH H OH HO H HO H H OH HO H H OH HO H H OH H OH H OH H OH CH OH CH2OH 2 CH2OH CH OH 2 (f) L-Aldohexose 8 possible CHO CHO CHO CHO CHO CHO CHO CHO H OH H OH H OH H OH HO H HO H HO H HO H H OH H OH HO H HO H H OH H OH HO H HO H H OH HO H H OH HO H H OH HO H H OH HO H HO H HO H HO H HO H HO H HO H HO H HO H CH2OH CH2OH CH2OH CH2OH CH2OH CH2OH CH2OH CH2OH 2. -

Stereochemistry Three-Dimensional Arrangement of Atoms (Groups) in Space
Chapter 9: Stereochemistry three-dimensional arrangement of atoms (groups) in space Stereoisomers: molecules with the same connectivity but different arrangement of atoms (groups) in space H C CH 3 3 H3C H H H H CH3 cis-1,2-dimethylcyclopropane trans-1,2-dimethylcyclopropane H H H CH3 H3C CH3 H3C H cis-2-butene trans-2-butene geometric isomers (diastereomers) 1 Enantiomers: non-superimposable mirror image isomers. Enantiomers are related to each other much like a right hand is related to a left hand Enantiomers have identical physical properties, i.e., bp, mp, etc. Chirality (from the Greek word for hand). Enantiomers are said to be chiral. Molecules are not chiral if they contain a plane of symmetry: a plane that cuts a molecule in half so that one half is the mirror image of the other half. Molecules (or objects) that possess a mirror plane of symmetry are superimposable on their mirror image and are termed achiral. A carbon with four different groups results in a chiral molecule and is refered to as a chiral or asymmetric or stereogenic center. 2 symmetry plane O H H H C O C C H Achiral H H O Not a symmetry plane O H H OH CH3 Chiral H C O C C H H H O Chiral center (stereogenic, asymmetric) Optical Rotation: molecules enriched in an enantiomer will rotate plane polarized light are are said to be optiically active. The optical rotation is dependent upon the substance, the concentration, the path length through the sample and the wavelength of light.