Flax Latitudinal Adaptation at Lutfl1 Altered Architecture and Promoted Fiber Production
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Checklist of the Vascular Plants of Redwood National Park
Humboldt State University Digital Commons @ Humboldt State University Botanical Studies Open Educational Resources and Data 9-17-2018 Checklist of the Vascular Plants of Redwood National Park James P. Smith Jr Humboldt State University, [email protected] Follow this and additional works at: https://digitalcommons.humboldt.edu/botany_jps Part of the Botany Commons Recommended Citation Smith, James P. Jr, "Checklist of the Vascular Plants of Redwood National Park" (2018). Botanical Studies. 85. https://digitalcommons.humboldt.edu/botany_jps/85 This Flora of Northwest California-Checklists of Local Sites is brought to you for free and open access by the Open Educational Resources and Data at Digital Commons @ Humboldt State University. It has been accepted for inclusion in Botanical Studies by an authorized administrator of Digital Commons @ Humboldt State University. For more information, please contact [email protected]. A CHECKLIST OF THE VASCULAR PLANTS OF THE REDWOOD NATIONAL & STATE PARKS James P. Smith, Jr. Professor Emeritus of Botany Department of Biological Sciences Humboldt State Univerity Arcata, California 14 September 2018 The Redwood National and State Parks are located in Del Norte and Humboldt counties in coastal northwestern California. The national park was F E R N S established in 1968. In 1994, a cooperative agreement with the California Department of Parks and Recreation added Del Norte Coast, Prairie Creek, Athyriaceae – Lady Fern Family and Jedediah Smith Redwoods state parks to form a single administrative Athyrium filix-femina var. cyclosporum • northwestern lady fern unit. Together they comprise about 133,000 acres (540 km2), including 37 miles of coast line. Almost half of the remaining old growth redwood forests Blechnaceae – Deer Fern Family are protected in these four parks. -
Plant List for Web Page
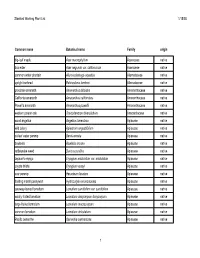
Stanford Working Plant List 1/15/08 Common name Botanical name Family origin big-leaf maple Acer macrophyllum Aceraceae native box elder Acer negundo var. californicum Aceraceae native common water plantain Alisma plantago-aquatica Alismataceae native upright burhead Echinodorus berteroi Alismataceae native prostrate amaranth Amaranthus blitoides Amaranthaceae native California amaranth Amaranthus californicus Amaranthaceae native Powell's amaranth Amaranthus powellii Amaranthaceae native western poison oak Toxicodendron diversilobum Anacardiaceae native wood angelica Angelica tomentosa Apiaceae native wild celery Apiastrum angustifolium Apiaceae native cutleaf water parsnip Berula erecta Apiaceae native bowlesia Bowlesia incana Apiaceae native rattlesnake weed Daucus pusillus Apiaceae native Jepson's eryngo Eryngium aristulatum var. aristulatum Apiaceae native coyote thistle Eryngium vaseyi Apiaceae native cow parsnip Heracleum lanatum Apiaceae native floating marsh pennywort Hydrocotyle ranunculoides Apiaceae native caraway-leaved lomatium Lomatium caruifolium var. caruifolium Apiaceae native woolly-fruited lomatium Lomatium dasycarpum dasycarpum Apiaceae native large-fruited lomatium Lomatium macrocarpum Apiaceae native common lomatium Lomatium utriculatum Apiaceae native Pacific oenanthe Oenanthe sarmentosa Apiaceae native 1 Stanford Working Plant List 1/15/08 wood sweet cicely Osmorhiza berteroi Apiaceae native mountain sweet cicely Osmorhiza chilensis Apiaceae native Gairdner's yampah (List 4) Perideridia gairdneri gairdneri Apiaceae -

Molecular Genetic Studies in Flax (Linum Usitatissimum L.)
Molecular genetic studies in flax (Linum usitatissimum L.) Moleculair genetische studies in vlas (Linum usitatissimum L.) Promotor: Prof. dr. ir. P. Stam Hoogleraar in de Plantenveredeling Copromotor: Dr. ir. H.J. van Eck Universitair docent, Laboratorium voor plantenveredeling Promotiecommissie: Prof. dr. A.G.M. Gerats (Radboud Universiteit Nijmegen) Prof. dr. R.F. Hoekstra (Wageningen Universiteit) Dr. R.G. van den Berg (Wageningen Universiteit) Dr. ir. J.W. van Ooijen (Kyazma B.V., Wageningen) Dit onderzoek is uitgevoerd binnen de onderzoeksscholen ‘Experimental Plant Sciences’ en ‘Production Ecology and Resource Conservation’. Molecular genetic studies in flax (Linum usitatissimum L.) Jaap Vromans Proefschrift ter verkrijging van de graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof. Dr. M.J. Kropff, in het openbaar te verdedigen op woensdag 15 maart 2006 des namiddags te vier uur in de Aula CIP-DTA Koninklijke Bibliotheek, Den Haag Molecular genetic studies in flax (Linum usitatissimum L.) Jaap Vromans PhD thesis, Wageningen University, The Netherlands With references – with summaries in Dutch and English ISBN 90-8504-374-3 Contents Chapter 1 General introduction 7 Chapter 2 Molecular analysis of flax (Linum usitatissimum L.) reveals a narrow genetic basis of fiber flax 21 Chapter 3 The molecular genetic variation in the genus Linum 41 Chapter 4 Selection of highly polymorphic AFLP fingerprints in flax (Linum usitatissimum L.) reduces the workload in genetic linkage map construction and results in -

Flora Mediterranea 26
FLORA MEDITERRANEA 26 Published under the auspices of OPTIMA by the Herbarium Mediterraneum Panormitanum Palermo – 2016 FLORA MEDITERRANEA Edited on behalf of the International Foundation pro Herbario Mediterraneo by Francesco M. Raimondo, Werner Greuter & Gianniantonio Domina Editorial board G. Domina (Palermo), F. Garbari (Pisa), W. Greuter (Berlin), S. L. Jury (Reading), G. Kamari (Patras), P. Mazzola (Palermo), S. Pignatti (Roma), F. M. Raimondo (Palermo), C. Salmeri (Palermo), B. Valdés (Sevilla), G. Venturella (Palermo). Advisory Committee P. V. Arrigoni (Firenze) P. Küpfer (Neuchatel) H. M. Burdet (Genève) J. Mathez (Montpellier) A. Carapezza (Palermo) G. Moggi (Firenze) C. D. K. Cook (Zurich) E. Nardi (Firenze) R. Courtecuisse (Lille) P. L. Nimis (Trieste) V. Demoulin (Liège) D. Phitos (Patras) F. Ehrendorfer (Wien) L. Poldini (Trieste) M. Erben (Munchen) R. M. Ros Espín (Murcia) G. Giaccone (Catania) A. Strid (Copenhagen) V. H. Heywood (Reading) B. Zimmer (Berlin) Editorial Office Editorial assistance: A. M. Mannino Editorial secretariat: V. Spadaro & P. Campisi Layout & Tecnical editing: E. Di Gristina & F. La Sorte Design: V. Magro & L. C. Raimondo Redazione di "Flora Mediterranea" Herbarium Mediterraneum Panormitanum, Università di Palermo Via Lincoln, 2 I-90133 Palermo, Italy [email protected] Printed by Luxograph s.r.l., Piazza Bartolomeo da Messina, 2/E - Palermo Registration at Tribunale di Palermo, no. 27 of 12 July 1991 ISSN: 1120-4052 printed, 2240-4538 online DOI: 10.7320/FlMedit26.001 Copyright © by International Foundation pro Herbario Mediterraneo, Palermo Contents V. Hugonnot & L. Chavoutier: A modern record of one of the rarest European mosses, Ptychomitrium incurvum (Ptychomitriaceae), in Eastern Pyrenees, France . 5 P. Chène, M. -

Vascular Plants of Humboldt Bay's Dunes and Wetlands Published by U.S
Vascular Plants of Humboldt Bay's Dunes and Wetlands Published by U.S. Fish and Wildlife Service G. Leppig and A. Pickart and California Department of Fish Game Release 4.0 June 2014* www.fws.gov/refuge/humboldt_bay/ Habitat- Habitat - Occurs on Species Status Occurs within Synonyms Common name specific broad Lanphere- Jepson Manual (2012) (see codes at end) refuge (see codes at end) (see codes at end) Ma-le'l Units UD PW EW Adoxaceae Sambucus racemosa L. red elderberry RF, CDF, FS X X N X X Aizoaceae Carpobrotus chilensis (Molina) sea fig DM X E X X N.E. Br. Carpobrotus edulis ( L.) N.E. Br. Iceplant DM X E, I X Alismataceae lanceleaf water Alisma lanceolatum With. FM X E plantain northern water Alisma triviale Pursh FM X N plantain Alliaceae three-cornered Allium triquetrum L. FS, FM, DM X X E leek Allium unifolium Kellogg one-leaf onion CDF X N X X Amaryllidaceae Amaryllis belladonna L. belladonna lily DS, AW X X E Narcissus pseudonarcissus L. daffodil AW, DS, SW X X E X Anacardiaceae Toxicodendron diversilobum Torrey poison oak CDF, RF X X N X X & A. Gray (E. Greene) Apiaceae Angelica lucida L. seacoast angelica BM X X N, C X X Anthriscus caucalis M. Bieb bur chevril DM X E Cicuta douglasii (DC.) J. Coulter & western water FM X N Rose hemlock Conium maculatum L. poison hemlock RF, AW X I X Daucus carota L. Queen Anne's lace AW, DM X X I X American wild Daucus pusillus Michaux DM, SW X X N X X carrot Foeniculum vulgare Miller sweet fennel AW, FM, SW X X I X Glehnia littoralis (A. -

Genome-Wide Identification, Characterization, and Expression
www.nature.com/scientificreports OPEN Genome-wide identification, characterization, and expression profile of aquaporin gene family in Received: 23 August 2016 Accepted: 13 March 2017 flax (Linum usitatissimum) Published: 27 April 2017 S. M. Shivaraj1,*, Rupesh K. Deshmukh2,*, Rhitu Rai1, Richard Bélanger2, Pawan K. Agrawal3 & Prasanta K. Dash1 Membrane intrinsic proteins (MIPs) form transmembrane channels and facilitate transport of myriad substrates across the cell membrane in many organisms. Majority of plant MIPs have water transporting ability and are commonly referred as aquaporins (AQPs). In the present study, we identified aquaporin coding genes in flax by genome-wide analysis, their structure, function and expression pattern by pan- genome exploration. Cross-genera phylogenetic analysis with known aquaporins from rice, arabidopsis, and poplar showed five subgroups of flax aquaporins representing 16 plasma membrane intrinsic proteins (PIPs), 17 tonoplast intrinsic proteins (TIPs), 13 NOD26-like intrinsic proteins (NIPs), 2 small basic intrinsic proteins (SIPs), and 3 uncharacterized intrinsic proteins (XIPs). Amongst aquaporins, PIPs contained hydrophilic aromatic arginine (ar/R) selective filter but TIP, NIP, SIP and XIP subfamilies mostly contained hydrophobic ar/R selective filter. Analysis of RNA-seq and microarray data revealed high expression of PIPs in multiple tissues, low expression of NIPs, and seed specific expression of TIP3 in flax. Exploration of aquaporin homologs in three closely relatedLinum species bienne, grandiflorum and leonii revealed presence of 49, 39 and 19 AQPs, respectively. The genome-wide identification of aquaporins, first in flax, provides insight to elucidate their physiological and developmental roles in flax. Water absorption from soil through roots and its translocation to different parts is of paramount importance for innate physiological processes in plants. -

) 2 10( ;3 201 Life Science Journal 659
Science Journal 210(;3201Life ) http://www.lifesciencesite.com Habitats and plant diversity of Al Mansora and Jarjr-oma regions in Al- Jabal Al- Akhdar- Libya Abusaief, H. M. A. Agron. Depar. Fac. Agric., Omar Al-Mukhtar Univ. [email protected] Abstract: Study conducted in two areas of Al Mansora and Jarjr-oma regions in Al- Jabal Al- Akhdar on the coast. The Rocky habitat Al Mansora 6.5 km of the Mediterranean Sea with altitude at 309.4 m, distance Jarjr-oma 300 m of the sea with altitude 1 m and distance. Vegetation study was undertaken during the autumn 2010 and winter, spring and summer 2011. The applied classification technique was the TWINSPAN, Divided ecologically into six main habitats to the vegetation in Rocky habitat of Al Mansora and five habitats in Jarjr oma into groups depending on the average number of species in habitats and community: In Rocky habitat Al Mansora community vegetation type Cistus parviflorus, Erica multiflora, Teucrium apollinis, Thymus capitatus, Micromeria Juliana, Colchium palaestinum and Arisarum vulgare. In Jarjr oma existed five habitat Salt march habitat Community dominant species by Suaeda vera, Saline habitat species Onopordum cyrenaicum, Rocky coastal habitat species Rumex bucephalophorus, Sandy beach habitat species Tamarix tetragyna and Sand formation habitat dominant by Retama raetem. The number of species in the Rocky habitat Al Mansora 175 species while in Jarjr oma reached 19 species of Salt march habitat and Saline habitat 111 species and 153 of the Rocky coastal habitat and reached to 33 species in Sandy beach and 8 species of Sand formations habitat. -

Diversity and Evolution of Rosids
Oxalidales • small, heterogeneous, novel group Diversity and of 6 families - seed character? Oxalidaceae Evolution of Rosids Wood sorrels . violets, willows, and spurges . Cephalotaceae Australian pitcher plant Oxalidaceae - wood sorrels Oxalidaceae - wood sorrels 6 genera, 770 species in the tropics and temperate areas - 700 6 genera, 770 species in the tropics and temperate areas - 700 belong to Oxalis (wood sorrel) belong to Oxalis (wood sorrel) • plants are herbaceous creepers or woody Oxalis corniculata - creeping yellow wood sorrel • typically 3-foliate vines leaves (the real shamrock) • leaves are acidic to taste due to oxalic acid in the form of calcium oxalate Oxalidaceae - wood sorrels Oxalidaceae - wood sorrels CA 5 CO 5 A 5+5 G (5) • 5 merous flowers CA 5 CO 5 A 5+5 G (5) • 5 merous flowers Oxalis corniculata Oxalis • fruits are 5 locular & Oxalis corniculata Oxalis • fruits are 5 locular & winged capsules or berries winged capsules or berries • tristyly common (3 levels at which 2 sets of anthers and 1 set of styles position) U U U Oxalidaceae - wood sorrels Oxalidaceae - wood sorrels • common native and introduced wood-sorrels • tropical fruit - carambola or star fruit: note 5 carpellate structure Oxalis stricta - Oxalis violaceae - tall wood-sorrel violet wood-sorrel Averrhoa carambola Oxalis acetosella - wood-sorrel *Malpighiales *Malpighiales • large and diverse group of 38 • unresolved! “novel” clade families - many of them • leaf margin teeth contributing importantly to tropical • “Parietales” subclade (placentation) forest diversity • hosts for Cymothoe butterflies *Malpighiales *Violaceae - violets • unusual life forms 23 genera, 800 species of herbs (temperate) to vines and small trees (tropics). 400-600 of them are violets (Viola). -

Pleistocene Radiation of the Serpentine-Adapted Genus Hesperolinon and Other Divergence Times in Linaceae (Malpighiales)
UC Berkeley UC Berkeley Previously Published Works Title Pleistocene radiation of the serpentine-adapted genus Hesperolinon and other divergence times in Linaceae (Malpighiales). Permalink https://escholarship.org/uc/item/6mx3r5r6 Journal American journal of botany, 103(2) ISSN 0002-9122 Authors Schneider, Adam C Freyman, William A Guilliams, C Matt et al. Publication Date 2016-02-05 DOI 10.3732/ajb.1500379 Peer reviewed eScholarship.org Powered by the California Digital Library University of California RESEARCH ARTICLE AMERICAN JOURNAL OF BOTANY Pleistocene radiation of the serpentine-adapted genus Hesperolinon and other divergence times in Linaceae (Malpighiales) 1 Adam C. Schneider 2,5 , William A. Freyman 2 , C. Matt Guilliams 2,3 , Yuri P. Springer 4 , and Bruce G. Baldwin 2 PREMISE OF THE STUDY: Hesperolinon (western fl ax; Linaceae) is endemic to the western United States, where it is notable for its high and geographically concentrated species diversity on serpentine-derived soils and for its use as a model system in disease ecology. We used a phylogenetic framework to test a long-standing hypothesis that Hesperolinon is a neoendemic radiation. METHODS: Five plastid and two ribosomal nuclear DNA gene regions were sampled from 105 populations of Hesperolinon , including all 13 recently recog- nized species across their known ranges. We used these data to generate population-level phylogenies of Hesperolinon . We also generated a robustly sampled chronogram of Linaceae using an eight-gene, 100-taxon supermatrix calibrated using fossil Linum pollen and a published chronogram of Malpighiales. KEY RESULTS: Most diversifi cation in Hesperolinon has taken place in the past 1–2 million yr, much more recently than previous estimates. -

Plant Paleopolyploidy James C
Plant Paleopolyploidy James C. Schnable1 and Eric Lyons3,4 1Center for Plant Science Innovation, University of Nebraska-Lincoln, Lincoln, NE, USA 2Department of Agronomy and Horticulture, University of Nebraska-Lincoln, Lincoln, NE, USA 3School of Plant Sciences, University of Arizona, Tucson, AZ, USA 4iPlant Collaborative, Bio5 Institute, University of Arizona, Tucson, AZ, USA ABSTRACT Identifying and characterizing plant paleopolyploidies is remains an ongoing area of investigation. Ancient whole genome duplications can be identified through whole genome comparisons using a combination of the data derived from genomic structure (e.g. syntenic dotplots) and evolutionary distances (e.g. synonymous mutation rates). These methods require large genomic and/or transcriptomic datasets, and our understanding identity, placement, and timing of these ancient events continues to change as new datasets become available. The images and texzt presented here represent our best estimates using the data available at this time, but, like many things, must be seen as tentative and subject to revision as new datasets and types of analyses become available. Keywords: Comparative Genomics, Plant Comparative Genomics, Whole Genome Duplication, Plant Paleopolyploidy INTRODUCTION Figure 1. Phylogenetic tree of plant species, predominantly those with published genome sequences with known ancient whole genome duplications marked. Tree is current as of April 2013. 1 EUDICOT HEXAPLOIDY Synonyms: Arabidopsis Gamm This hexaploidy (genome tripling) is shared by the core eudicots (the rosids and asterids), and may be present in additional, basal eudicots, although it will not be possible to reach this conclusion until the genomes of species from basal lineages are sequenced and assembled to the pseudomolecule level. This whole genome duplication was first identified as the most ancient of three whole genome duplication present in the genome of Arabidopsis thaliana, and assigned the name ”gamma” by in 2003 by (Bowers et al., 2003). -

Vascular Plants of Trinidad Head, Humboldt County, California
Humboldt State University Digital Commons @ Humboldt State University Botanical Studies Open Educational Resources and Data 2017 Vascular Plants of Trinidad Head, Humboldt County, California James P. Smith Jr Humboldt State University, [email protected] Follow this and additional works at: https://digitalcommons.humboldt.edu/botany_jps Part of the Botany Commons Recommended Citation Smith, James P. Jr, "Vascular Plants of Trinidad Head, Humboldt County, California" (2017). Botanical Studies. 50. https://digitalcommons.humboldt.edu/botany_jps/50 This Flora of Northwest California-Checklists of Local Sites is brought to you for free and open access by the Open Educational Resources and Data at Digital Commons @ Humboldt State University. It has been accepted for inclusion in Botanical Studies by an authorized administrator of Digital Commons @ Humboldt State University. For more information, please contact [email protected]. THE VASCULAR PLANTS OF TRINIDAD HEAD, HUMBOLDT COUNTY, CALIFORNIA James P. Smith, Jr. Professor Emeritus of Botany Department of Biological Sciences Humboldt State University Arcata, California 3 June 2017 Trinidad Head is located 1.5 miles southwest of the City of Trinidad (N 41.0541, W -124.1505). It is composed C O N I F E R S of igneous rock surrounded by a Franciscan melange, with Pleistocene sands and gravel on top. CUPRESSACEAE • CYPRESS FAMILY Sequoia sempervirens • coast redwood Trinidad Head is a sacred site for the Yurok People. It was later claimed for Spain in 1775 by Bruno de Heceta. PINACEAE • PINE FAMILY It is the site of a global baseline atmospheric Abies grandis • grand fir observatory operated by NOAA. In January 2017, Picea sitchensis • Sitka spruce President Obama added Trinidad Head to an expanded Pinus contorta • beach pine California Coastal National Monument. -

Salix, Willow 154 1
on biodiversity and geneflow of selected biofuel crops Klaus Ammann Delft University of Technology, [email protected] January - February 2007, 10 studies for the working groups of www.epobio.net Avena, Beta vulgaris, Brassica napus, Crambe, Linum, Miscanthus, Nicotiana tabaccum, Populus, Salix, Triticum 2 Contents Contents ............................................................................................................................................. 2 Figures................................................................................................................................................ 4 Avena, Oats 11 1. Taxonomy ..................................................................................................................................... 11 2. Biosafety considerations............................................................................................................... 14 3. Transgenic oats ............................................................................................................................ 16 4. Management and mitigation of gene flow..................................................................................... 20 Gene Flow Assessment for Avena ................................................................................................... 26 Beta vulgaris, Beet 28 1. Taxonomy ..................................................................................................................................... 29 2. Reproduction biology...................................................................................................................