Durham E-Theses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pollinator Niche Partitioning and Asymmetric Facilitation Contribute to The
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.02.974022; this version posted March 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 1 Pollinator niche partitioning and asymmetric facilitation contribute to the 2 maintenance of diversity 3 Na Wei,1,2* Rainee L. Kaczorowski,1 Gerardo Arceo-Gómez,1,3 Elizabeth M. O'Neill,1 Rebecca 4 A. Hayes,1 Tia-Lynn Ashman1* 5 Affiliations: 6 1Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA, USA. 7 2The Holden Arboretum, Kirtland, OH, USA. 8 3Department of Biological Sciences, East Tennessee State University, Johnson City, TN, USA. 9 *Corresponding authors. Email: [email protected] and [email protected]. 10 Abstract: 11 Mechanisms that favor rare species are key to the maintenance of diversity. One of the most 12 critical tasks for biodiversity conservation is understanding how plant–pollinator mutualisms 13 contribute to the persistence of rare species, yet this remains poorly understood. Using a process- 14 based model that integrates plant–pollinator and interspecific pollen transfer networks with floral 15 functional traits, we show that niche partitioning in pollinator use and asymmetric facilitation 16 confer fitness advantage of rare species in a biodiversity hotspot. While co-flowering species 17 filtered pollinators via floral traits, rare species showed greater pollinator specialization leading 18 to higher pollination-mediated male and female fitness than abundant species. When plants 19 shared pollinator resources, asymmetric facilitation via pollen transport dynamics benefited the 20 rare species at the cost of the abundant ones, serving as an alternative diversity-promoting 21 mechanism. -

FLORA from FĂRĂGĂU AREA (MUREŞ COUNTY) AS POTENTIAL SOURCE of MEDICINAL PLANTS Silvia OROIAN1*, Mihaela SĂMĂRGHIŢAN2
ISSN: 2601 – 6141, ISSN-L: 2601 – 6141 Acta Biologica Marisiensis 2018, 1(1): 60-70 ORIGINAL PAPER FLORA FROM FĂRĂGĂU AREA (MUREŞ COUNTY) AS POTENTIAL SOURCE OF MEDICINAL PLANTS Silvia OROIAN1*, Mihaela SĂMĂRGHIŢAN2 1Department of Pharmaceutical Botany, University of Medicine and Pharmacy of Tîrgu Mureş, Romania 2Mureş County Museum, Department of Natural Sciences, Tîrgu Mureş, Romania *Correspondence: Silvia OROIAN [email protected] Received: 2 July 2018; Accepted: 9 July 2018; Published: 15 July 2018 Abstract The aim of this study was to identify a potential source of medicinal plant from Transylvanian Plain. Also, the paper provides information about the hayfields floral richness, a great scientific value for Romania and Europe. The study of the flora was carried out in several stages: 2005-2008, 2013, 2017-2018. In the studied area, 397 taxa were identified, distributed in 82 families with therapeutic potential, represented by 164 medical taxa, 37 of them being in the European Pharmacopoeia 8.5. The study reveals that most plants contain: volatile oils (13.41%), tannins (12.19%), flavonoids (9.75%), mucilages (8.53%) etc. This plants can be used in the treatment of various human disorders: disorders of the digestive system, respiratory system, skin disorders, muscular and skeletal systems, genitourinary system, in gynaecological disorders, cardiovascular, and central nervous sistem disorders. In the study plants protected by law at European and national level were identified: Echium maculatum, Cephalaria radiata, Crambe tataria, Narcissus poeticus ssp. radiiflorus, Salvia nutans, Iris aphylla, Orchis morio, Orchis tridentata, Adonis vernalis, Dictamnus albus, Hammarbya paludosa etc. Keywords: Fărăgău, medicinal plants, human disease, Mureş County 1. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -

Two New Genera in the Omphalodes Group (Cynoglosseae, Boraginaceae)
Nova Acta Científica Compostelana (Bioloxía),23 : 1-14 (2016) - ISSN 1130-9717 ARTÍCULO DE INVESTIGACIÓN Two new genera in the Omphalodes group (Cynoglosseae, Boraginaceae) Dous novos xéneros no grupo Omphalodes (Cynoglosseae, Boraginaceae) M. SERRANO1, R. CARBAJAL1, A. PEREIRA COUTINHO2, S. ORTIZ1 1 Department of Botany, Faculty of Pharmacy, University of Santiago de Compostela, 15782 Santiago de Compostela , Spain 2 CFE, Centre for Functional Ecology, Department of Life Sciences, University of Coimbra, 3000-456 Coimbra, Portugal *[email protected]; [email protected]; [email protected]; [email protected] *: Corresponding author (Recibido: 08/06/2015; Aceptado: 01/02/2016; Publicado on-line: 04/02/2016) Abstract Omphalodes (Boraginaceae, Cynoglosseae) molecular phylogenetic relationships are surveyed in the context of the tribe Cynoglosseae, being confirmed that genusOmphalodes is paraphyletic. Our work is focused both in the internal relationships among representatives of Omphalodes main subgroups (and including Omphalodes verna, the type species), and their relationships with other Cynoglosseae genera that have been related to the Omphalodes group. Our phylogenetic analysis of ITS and trnL-trnF molecular markers establish close relationships of the American Omphalodes with the genus Mimophytum, and also with Cynoglossum paniculatum and Myosotidium hortensia. The southwestern European annual Omphalodes species form a discrete group deserving taxonomic recognition. We describe two new genera to reduce the paraphyly in the genus Omphalodes, accommodating the European annual species in Iberodes and Cynoglossum paniculatum in Mapuchea. The pollen of the former taxon is described in detail for the first time. Keywords: Madrean-Tethyan, phylogeny, pollen, systematics, taxonomy Resumo Neste estudo analisamos as relacións filoxenéticas deOmphalodes (Boraginaceae, Cynoglosseae) no contexto da tribo Cynoglosseae, confirmándose como parafilético o xéneroOmphalodes . -

Anti-HIV-1 Activities of the Extracts from the Medicinal Plant Linum Grandiflorum Desf
® Medicinal and Aromatic Plant Science and Biotechnology ©2009 Global Science Books Anti-HIV-1 Activities of the Extracts from the Medicinal Plant Linum grandiflorum Desf. Magdy M. D. Mohammed1,2,3* • Lars P. Christensen1,4 • Nabaweya A. Ibrahim3 • Nagwa E. Awad3 • Ibrahim F. Zeid5 • Erik B. Pedersen2 • Kenneth B. Jensen2 • Paolo L. Colla6 1 Danish Institute of Agricultural Sciences (DIAS), Department of Food Science, Research Center Aarslev, Aarhus University, Kirstinebjergvej 10, DK-5792 Aarslev, Denmark 2 Nucleic Acid Center, Institute of Physics and Chemistry, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark 3 Pharmacognosy Department, National Research Center, Dokki-12311, Cairo, Egypt 4 Institute of Chemical Engineering, Biotechnology and Environmental Technology, University of Southern Denmark, Niels Bohrs Allé 1, DK-5230 Odense M, Denmark 5 Chemistry Department, Faculty of Science, El-Menoufia University, El-Menoufia, Egypt 6 Department of Science and Biomedical Technology, Cittadella University, Monserrato, Italy Corresponding author : * [email protected] ABSTRACT As part of our screening of anti-AIDS agents from natural sources e.g. Ixora undulata, Paulownia tomentosa, Fortunella margarita, Aegle marmelos and Erythrina abyssinica, the different organic and aqueous extracts of Linum grandiflorum leaves and seeds were evaluated in vitro by the microculture tetrazolium (MTT) assay. The activity of the tested extracts against multiplication of HIV-1 wild type IIIB, N119, A17, and EFVR in acutely infected cells was based on inhibition of virus-induced cytopathicity in MT-4 cells. Results revealed that both the MeOH and the CHCl3 extracts of L. grandiflorum have significant inhibitory effects against HIV-1 induced infection with MT-4 cells. -

Checklist of the Vascular Plants of Redwood National Park
Humboldt State University Digital Commons @ Humboldt State University Botanical Studies Open Educational Resources and Data 9-17-2018 Checklist of the Vascular Plants of Redwood National Park James P. Smith Jr Humboldt State University, [email protected] Follow this and additional works at: https://digitalcommons.humboldt.edu/botany_jps Part of the Botany Commons Recommended Citation Smith, James P. Jr, "Checklist of the Vascular Plants of Redwood National Park" (2018). Botanical Studies. 85. https://digitalcommons.humboldt.edu/botany_jps/85 This Flora of Northwest California-Checklists of Local Sites is brought to you for free and open access by the Open Educational Resources and Data at Digital Commons @ Humboldt State University. It has been accepted for inclusion in Botanical Studies by an authorized administrator of Digital Commons @ Humboldt State University. For more information, please contact [email protected]. A CHECKLIST OF THE VASCULAR PLANTS OF THE REDWOOD NATIONAL & STATE PARKS James P. Smith, Jr. Professor Emeritus of Botany Department of Biological Sciences Humboldt State Univerity Arcata, California 14 September 2018 The Redwood National and State Parks are located in Del Norte and Humboldt counties in coastal northwestern California. The national park was F E R N S established in 1968. In 1994, a cooperative agreement with the California Department of Parks and Recreation added Del Norte Coast, Prairie Creek, Athyriaceae – Lady Fern Family and Jedediah Smith Redwoods state parks to form a single administrative Athyrium filix-femina var. cyclosporum • northwestern lady fern unit. Together they comprise about 133,000 acres (540 km2), including 37 miles of coast line. Almost half of the remaining old growth redwood forests Blechnaceae – Deer Fern Family are protected in these four parks. -
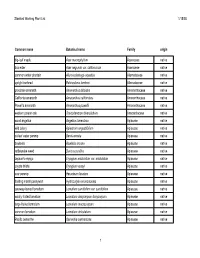
Plant List for Web Page
Stanford Working Plant List 1/15/08 Common name Botanical name Family origin big-leaf maple Acer macrophyllum Aceraceae native box elder Acer negundo var. californicum Aceraceae native common water plantain Alisma plantago-aquatica Alismataceae native upright burhead Echinodorus berteroi Alismataceae native prostrate amaranth Amaranthus blitoides Amaranthaceae native California amaranth Amaranthus californicus Amaranthaceae native Powell's amaranth Amaranthus powellii Amaranthaceae native western poison oak Toxicodendron diversilobum Anacardiaceae native wood angelica Angelica tomentosa Apiaceae native wild celery Apiastrum angustifolium Apiaceae native cutleaf water parsnip Berula erecta Apiaceae native bowlesia Bowlesia incana Apiaceae native rattlesnake weed Daucus pusillus Apiaceae native Jepson's eryngo Eryngium aristulatum var. aristulatum Apiaceae native coyote thistle Eryngium vaseyi Apiaceae native cow parsnip Heracleum lanatum Apiaceae native floating marsh pennywort Hydrocotyle ranunculoides Apiaceae native caraway-leaved lomatium Lomatium caruifolium var. caruifolium Apiaceae native woolly-fruited lomatium Lomatium dasycarpum dasycarpum Apiaceae native large-fruited lomatium Lomatium macrocarpum Apiaceae native common lomatium Lomatium utriculatum Apiaceae native Pacific oenanthe Oenanthe sarmentosa Apiaceae native 1 Stanford Working Plant List 1/15/08 wood sweet cicely Osmorhiza berteroi Apiaceae native mountain sweet cicely Osmorhiza chilensis Apiaceae native Gairdner's yampah (List 4) Perideridia gairdneri gairdneri Apiaceae -

Curriculum Vitae Adam Schneider
Curriculum Vitae Adam Schneider Department of Integrative Biology and (608) 445-9083 University and Jepson Herbaria [email protected] University of California – Berkeley 1001 Valley Life Sciences Building Berkeley, CA 94720-2465 Education Ph.D. Candidate, Integrative Biology University of California–Berkeley Major Advisor: Bruce Baldwin Advanced to Candidacy May 2014 B.S. Biology, Chemistry University of Wisconsin–Eau Claire (Summa Cum Laude) 2012 International Student University of Ghana–Legon Spring 2011 Professional Appointments and Fellowships University of California, Berkeley Berkeley Graduate Fellow 2012-present Graduate Student Researcher 2013-2015 Graduate Student Instructor 2013-2015 Taught laboratory, field trips, and guest lectures for: California Plant Life (2 sem.) Medical Ethnobotany (2 sem. + summer) Plant Systematics (1 sem.) Charles Darwin Research Station Herbarium, Galápagos, Ecuador Curatorial Intern May-Aug 2011 University of Wisconsin, Eau Claire Blugold Fellow 2009- 2011 - Planned and executed a study of plant responses to differing water regimes with Dr. Tali Lee. Publications In Review Schneider A.C., W. A. Freyman, C. M. Guilliams, Y. P. Springer, B. G. Baldwin (in review) Pleistocene radiation of the serpentine-adapted genus Hesperolinon and other divergence times in Linaceae (Malpighiales). American Journal of Botany. Kulhanek K.A.*, L.C. Ponicio*, A.C. Schneider*, R. E. Walsh* (in review) Strategic conversation: Mission and relevance of national parks. In Beissinger S.R., D.D. Ackerly, H. Dormus, G. Machelis, Science for parks, parks for science: The next century. University of Chicago Press. Curriculum Vitae Adam Schneider Page 2 Peer-reviewed Publications: 1. Schneider, A.C., T.D. Lee, M.A. Kreiser, G.T. -

Vegetative Growth and Organogenesis 555
Vegetative Growth 19 and Organogenesis lthough embryogenesis and seedling establishment play criti- A cal roles in establishing the basic polarity and growth axes of the plant, many other aspects of plant form reflect developmental processes that occur after seedling establishment. For most plants, shoot architecture depends critically on the regulated production of determinate lateral organs, such as leaves, as well as the regulated formation and outgrowth of indeterminate branch systems. Root systems, though typically hidden from view, have comparable levels of complexity that result from the regulated formation and out- growth of indeterminate lateral roots (see Chapter 18). In addition, secondary growth is the defining feature of the vegetative growth of woody perennials, providing the structural support that enables trees to attain great heights. In this chapter we will consider the molecular mechanisms that underpin these growth patterns. Like embryogenesis, vegetative organogenesis and secondary growth rely on local differences in the interactions and regulatory feedback among hormones, which trigger complex programs of gene expres- sion that drive specific aspects of organ development. Leaf Development Morphologically, the leaf is the most variable of all the plant organs. The collective term for any type of leaf on a plant, including struc- tures that evolved from leaves, is phyllome. Phyllomes include the photosynthetic foliage leaves (what we usually mean by “leaves”), protective bud scales, bracts (leaves associated with inflorescences, or flowers), and floral organs. In angiosperms, the main part of the foliage leaf is expanded into a flattened structure, the blade, or lamina. The appearance of a flat lamina in seed plants in the middle to late Devonian was a key event in leaf evolution. -

Honey Bee Suite © Rusty Burlew 2015 Master Plant List by Scientific Name United States
Honey Bee Suite Master Plant List by Scientific Name United States © Rusty Burlew 2015 Scientific name Common Name Type of plant Zone Full Link for more information Abelia grandiflora Glossy abelia Shrub 6-9 http://plants.ces.ncsu.edu/plants/all/abelia-x-grandiflora/ Acacia Acacia Thorntree Tree 3-8 http://www.2020site.org/trees/acacia.html Acer circinatum Vine maple Tree 7-8 http://www.nwplants.com/business/catalog/ace_cir.html Acer macrophyllum Bigleaf maple Tree 5-9 http://treesandshrubs.about.com/od/commontrees/p/Big-Leaf-Maple-Acer-macrophyllum.htm Acer negundo L. Box elder Tree 2-10 http://www.missouribotanicalgarden.org/PlantFinder/PlantFinderDetails.aspx?kempercode=a841 Acer rubrum Red maple Tree 3-9 http://www.missouribotanicalgarden.org/PlantFinder/PlantFinderDetails.aspx?taxonid=275374&isprofile=1&basic=Acer%20rubrum Acer rubrum Swamp maple Tree 3-9 http://www.missouribotanicalgarden.org/PlantFinder/PlantFinderDetails.aspx?taxonid=275374&isprofile=1&basic=Acer%20rubrum Acer saccharinum Silver maple Tree 3-9 http://en.wikipedia.org/wiki/Acer_saccharinum Acer spp. Maple Tree 3-8 http://en.wikipedia.org/wiki/Maple Achillea millefolium Yarrow Perennial 3-9 http://www.missouribotanicalgarden.org/PlantFinder/PlantFinderDetails.aspx?kempercode=b282 Aesclepias tuberosa Butterfly weed Perennial 3-9 http://www.missouribotanicalgarden.org/PlantFinder/PlantFinderDetails.aspx?kempercode=b490 Aesculus glabra Buckeye Tree 3-7 http://www.missouribotanicalgarden.org/PlantFinder/PlantFinderDetails.aspx?taxonid=281045&isprofile=1&basic=buckeye -

Illustrated Flora of East Texas Illustrated Flora of East Texas
ILLUSTRATED FLORA OF EAST TEXAS ILLUSTRATED FLORA OF EAST TEXAS IS PUBLISHED WITH THE SUPPORT OF: MAJOR BENEFACTORS: DAVID GIBSON AND WILL CRENSHAW DISCOVERY FUND U.S. FISH AND WILDLIFE FOUNDATION (NATIONAL PARK SERVICE, USDA FOREST SERVICE) TEXAS PARKS AND WILDLIFE DEPARTMENT SCOTT AND STUART GENTLING BENEFACTORS: NEW DOROTHEA L. LEONHARDT FOUNDATION (ANDREA C. HARKINS) TEMPLE-INLAND FOUNDATION SUMMERLEE FOUNDATION AMON G. CARTER FOUNDATION ROBERT J. O’KENNON PEG & BEN KEITH DORA & GORDON SYLVESTER DAVID & SUE NIVENS NATIVE PLANT SOCIETY OF TEXAS DAVID & MARGARET BAMBERGER GORDON MAY & KAREN WILLIAMSON JACOB & TERESE HERSHEY FOUNDATION INSTITUTIONAL SUPPORT: AUSTIN COLLEGE BOTANICAL RESEARCH INSTITUTE OF TEXAS SID RICHARDSON CAREER DEVELOPMENT FUND OF AUSTIN COLLEGE II OTHER CONTRIBUTORS: ALLDREDGE, LINDA & JACK HOLLEMAN, W.B. PETRUS, ELAINE J. BATTERBAE, SUSAN ROBERTS HOLT, JEAN & DUNCAN PRITCHETT, MARY H. BECK, NELL HUBER, MARY MAUD PRICE, DIANE BECKELMAN, SARA HUDSON, JIM & YONIE PRUESS, WARREN W. BENDER, LYNNE HULTMARK, GORDON & SARAH ROACH, ELIZABETH M. & ALLEN BIBB, NATHAN & BETTIE HUSTON, MELIA ROEBUCK, RICK & VICKI BOSWORTH, TONY JACOBS, BONNIE & LOUIS ROGNLIE, GLORIA & ERIC BOTTONE, LAURA BURKS JAMES, ROI & DEANNA ROUSH, LUCY BROWN, LARRY E. JEFFORDS, RUSSELL M. ROWE, BRIAN BRUSER, III, MR. & MRS. HENRY JOHN, SUE & PHIL ROZELL, JIMMY BURT, HELEN W. JONES, MARY LOU SANDLIN, MIKE CAMPBELL, KATHERINE & CHARLES KAHLE, GAIL SANDLIN, MR. & MRS. WILLIAM CARR, WILLIAM R. KARGES, JOANN SATTERWHITE, BEN CLARY, KAREN KEITH, ELIZABETH & ERIC SCHOENFELD, CARL COCHRAN, JOYCE LANEY, ELEANOR W. SCHULTZE, BETTY DAHLBERG, WALTER G. LAUGHLIN, DR. JAMES E. SCHULZE, PETER & HELEN DALLAS CHAPTER-NPSOT LECHE, BEVERLY SENNHAUSER, KELLY S. DAMEWOOD, LOGAN & ELEANOR LEWIS, PATRICIA SERLING, STEVEN DAMUTH, STEVEN LIGGIO, JOE SHANNON, LEILA HOUSEMAN DAVIS, ELLEN D. -

Lilioceris Egena Air Potato Biocontrol Environmental Assessment
United States Department of Field Release of the Beetle Agriculture Lilioceris egena (Coleoptera: Marketing and Regulatory Chrysomelidae) for Classical Programs Biological Control of Air Potato, Dioscorea bulbifera (Dioscoreaceae), in the Continental United States Environmental Assessment, February 2021 Field Release of the Beetle Lilioceris egena (Coleoptera: Chrysomelidae) for Classical Biological Control of Air Potato, Dioscorea bulbifera (Dioscoreaceae), in the Continental United States Environmental Assessment, February 2021 Agency Contact: Colin D. Stewart, Assistant Director Pests, Pathogens, and Biocontrol Permits Plant Protection and Quarantine Animal and Plant Health Inspection Service U.S. Department of Agriculture 4700 River Rd., Unit 133 Riverdale, MD 20737 Non-Discrimination Policy The U.S. Department of Agriculture (USDA) prohibits discrimination against its customers, employees, and applicants for employment on the bases of race, color, national origin, age, disability, sex, gender identity, religion, reprisal, and where applicable, political beliefs, marital status, familial or parental status, sexual orientation, or all or part of an individual's income is derived from any public assistance program, or protected genetic information in employment or in any program or activity conducted or funded by the Department. (Not all prohibited bases will apply to all programs and/or employment activities.) To File an Employment Complaint If you wish to file an employment complaint, you must contact your agency's EEO Counselor (PDF) within 45 days of the date of the alleged discriminatory act, event, or in the case of a personnel action. Additional information can be found online at http://www.ascr.usda.gov/complaint_filing_file.html. To File a Program Complaint If you wish to file a Civil Rights program complaint of discrimination, complete the USDA Program Discrimination Complaint Form (PDF), found online at http://www.ascr.usda.gov/complaint_filing_cust.html, or at any USDA office, or call (866) 632-9992 to request the form.