Proteasome Subtypes and Regulators in the Processing of Antigenic Peptides Presented by Class I Molecules of the Major Histocompatibility Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antigen Presentation by Dendritic Cells and Their Significance in Antineoplastic Immunotherapy
in vivo 18: 81-100 (2004) Antigen Presentation by Dendritic Cells and their Significance in Antineoplastic Immunotherapy BELA BODEY1,2, STUART E. SIEGEL1,2 and HANS E. KAISER3 1Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA; 2Childrens’ Center for Cancer and Blood Diseases, Childrens’ Hospital Los Angeles, Los Angeles, CA; 3Department of Pathology, School of Medicine, University of Maryland, Baltimore, MD, U.S.A. and Department of General and Experimental Pathology, University of Vienna, Vienna, Austria Abstract. Dendritic cells (DCs) are present in essentially every derived peptides on the surface of major histocompatibility mammalian tissue, where they operate at the interface of innate complex (MHC) molecules and acquire the cellular specialization and acquired immunity by recognizing pathogens and presenting to select and activate naive antigen-specific T lymphocytes. pathogen-derived peptides to T lymphocytes. According to the Immunotherapeutic ideas are based on the ability of the research group of Shortman (1-9), experimental results suggest a mammalian immune system to recognize neoplastically "dual" DC differentiation model, demonstrating the existence of transformed cells. Immunotherapy of human neoplasms has both myeloid-derived (with characteristic IF: CD11b+, CD11c+, always represented a very attractive fourth-modality therapeutic CD8alpha- and DEC205+) and lymphoid-derived DCs (showing approach, especially in light of the many shortcomings of CD11b-, CD11c-, CD8alpha+ and DEC205+ IF). DCs, including conventional surgical, radiation and chemotherapies in the interdigitating cells (IDCs) and Langerhans cells (LCs), are management of neoplastically transformed cells. The cancer characterized by dendritic morphology, high migratory mobility and vaccine approach to therapy is based on the notion that the are the most effective, "professional" cells for antigen presentation immune system could possibly mount a rejection strength response in primary immune responses. -

Antigen Processing (Major Hbitocompatlbility Complex/Class I Moecules/Lymphokines) YOUNG YANG, JAMES B
Proc. Nati. Acad. Sci. USA Vol. 89, pp. 4928-4932, June 1992 Immunology Proteasomes are regulated by interferon y: Implications for antigen processing (major hbitocompatlbility complex/class I moecules/lymphokines) YOUNG YANG, JAMES B. WATERS, KLAUS FROH, AND PER A. PETERSON Department of Immunology, The Scripps Research Institute, La Jolla, CA 92037 Communicated by Frank J. Dixon, February 27, 1992 ABSTRACT Class I major histocompatibility complex strengthened by the findings, presented in this communica- (MHC) molecules present antigenic peptides of cytoplasmic tion, that several proteasomal subunits, including MHC- origin to T cells. As the lengths ofthese peptides seem stried encoded subunits, are regulated by interferon y (IFN--y) and to eight or nine amino acids, an unusual proteolytic system that the incorporation of several more subunits into protea- must play a role in antigen processing. Proteasomes, a major somes appears to depend on the expression of the MHC- extralysosomal proteolytic system, are responsible for the encoded proteasomal subunits. Moreover, the pattern of degradation of cytoplasmic proteins. We demonstrate that expression of IFN-y-regulated subunits suggests complexi- several proteasomal subunits, including MHC-encoded sub- ties in the regulation of proteasomes with respect to its units, are regulated by interferon y. These data and the finding subunit composition, subcellular localization, and its incor- that MHC-encoded and other interferon -regulated protea- poration into larger ubiquitin-related proteolytic complexes. somal subunits are uniquely associated with proteasomes Possible functions for the MHC-encoded and IFN-y- strongly suggest that the immune system has recruited protea- regulated proteasomal subunits in antigen processing are somes for antigen processing. -

Cytoplasmic Proteolysis Antigens on MHC Class II Is Dependent On
Efficient Presentation of Both Cytosolic and Endogenous Transmembrane Protein Antigens on MHC Class II Is Dependent on Cytoplasmic Proteolysis This information is current as of September 28, 2021. Paushali Mukherjee, Aadish Dani, Sumeena Bhatia, Nagendra Singh, Alexander Y. Rudensky, Anna George, Vineeta Bal, Satyajit Mayor and Satyajit Rath J Immunol 2001; 167:2632-2641; ; doi: 10.4049/jimmunol.167.5.2632 Downloaded from http://www.jimmunol.org/content/167/5/2632 References This article cites 51 articles, 20 of which you can access for free at: http://www.jimmunol.org/content/167/5/2632.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 28, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2001 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Efficient Presentation of Both Cytosolic and Endogenous Transmembrane Protein Antigens on MHC Class II Is Dependent on Cytoplasmic Proteolysis1 Paushali Mukherjee,* Aadish Dani,† Sumeena Bhatia,* Nagendra Singh,* Alexander Y. -
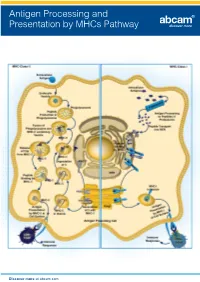
Antigen Processing and Presentation by Mhcs Pathway
Antigen Processing and Presentation by MHCs Pathway Discover more at abcam.com Antigen Processing and Presentation by MHCs Pathway Related antibodies Product This image shows MHC Class II highlights Product Clonality Applications Host Species Reactivity Product code expressing cells in formalin fixed and paraffin embedded human lymph MHC class I antibody [EP1395Y] M Flow Cyt, ICC/IF, IHC-P, IP, WB Rb Hu, Ms, Rat 52922 node, using ab55152. MHC class I antibody [AF6-88.5.5.3] (Biotin) M Flow Cyt Ms Ms 93528 MHC Class II antibody MHC class I antibody [34-1-2S] (FITC) M Flow Cyt Ms Ms 95572 (ab55152) MHC class I antibody [34-1-2S] (Phycoerythrin) M Flow Cyt Ms Ms 95571 MHC Class I alpha antibody [F21-2] M AP, Flow Cyt, IP, WB Ms Chk 23483 Clonality Applications MHC Class 1 H2 Db antibody [27-11-13] - BSA and Azide free M Flow Cyt, FuncS, IHC-Fr Ms Ms 25244 M IHC-P, WB MHC Class 1 H2 Db antibody [28-14-8] (FITC) M Flow Cyt Ms Ms 25056 Host Species cross reactivity MHC Class 1 H2 Db antibody [KH95] M Flow Cyt, FuncS, WB Ms Ms 64373 Ms Hu, RecFrag MHC Class I H2 Dd antibody [34-2-12] M Flow Cyt, FuncS, IHC-Fr, IP Ms Ms 64368 MHC Class I H2 Dk antibody [15-5-5.3] M Flow Cyt Ms Ms 25216 MHC Class II antigens are a valuable tool for studying T helper cell interactions with class II MHC Class I H2 Kb antibody P ELISA, WB Rb Ms 93364 positive antigen presenting cells, as well as the MHC Class I H2 Kd + Dd + q + u + v antibody [1.B.552] (FITC) M Flow Cyt, IP Ms Ms 62228 development of T helper cells since the antigen is present on stromal cells in the thymus. -

Mechanisms of HIV Protein Degradation Into Epitopes: Implications for Vaccine Design
Viruses 2014, 6, 3271-3292; doi:10.3390/v6083271 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Mechanisms of HIV Protein Degradation into Epitopes: Implications for Vaccine Design Marijana Rucevic †, Julie Boucau †, Jens Dinter †, Georgio Kourjian † and Sylvie Le Gall * Ragon Institute of MGH, MIT and Harvard, Massachusetts General Hospital and Harvard Medical School, Cambridge, MA 02139, USA; E-Mails: [email protected] (M.R.); [email protected] (J.B.); [email protected] (J.D.); [email protected] (G.K.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-857-268-7010. Received: 11 June 2014; in revised form: 6 August 2014 / Accepted: 11 August 2014 / Published: 21 August 2014 Abstract: The degradation of HIV-derived proteins into epitopes displayed by MHC-I or MHC-II are the first events leading to the priming of HIV-specific immune responses and to the recognition of infected cells. Despite a wealth of information about peptidases involved in protein degradation, our knowledge of epitope presentation during HIV infection remains limited. Here we review current data on HIV protein degradation linking epitope production and immunodominance, viral evolution and impaired epitope presentation. We propose that an in-depth understanding of HIV antigen processing and presentation in relevant primary cells could be exploited to identify signatures leading to efficient or inefficient epitope presentation in HIV proteomes, and to improve the design of immunogens eliciting immune responses efficiently recognizing all infected cells. Keywords: HIV; antigen processing; protein degradation; proteasome; aminopeptidase; peptidase; immunogen; vaccine vector; dendritic cells; T cells; viral evolution Viruses 2014, 6 3272 1. -

Footprints of Antigen Processing Boost MHC Class II Natural Ligand
Barra et al. Genome Medicine (2018) 10:84 https://doi.org/10.1186/s13073-018-0594-6 RESEARCH Open Access Footprints of antigen processing boost MHC class II natural ligand predictions Carolina Barra1† , Bruno Alvarez1†, Sinu Paul2, Alessandro Sette2, Bjoern Peters2, Massimo Andreatta1, Søren Buus3 and Morten Nielsen1,4* Abstract Background: Major histocompatibility complex class II (MHC-II) molecules present peptide fragments to T cells for immune recognition. Current predictors for peptide to MHC-II binding are trained on binding affinity data, generated in vitro and therefore lacking information about antigen processing. Methods: We generate prediction models of peptide to MHC-II binding trained with naturally eluted ligands derived from mass spectrometry in addition to peptide binding affinity data sets. Results: We show that integrated prediction models incorporate identifiable rules of antigen processing. In fact, we observed detectable signals of protease cleavage at defined positions of the ligands. We also hypothesize a role of the length of the terminal ligand protrusions for trimming the peptide to the MHC presented ligand. Conclusions: The results of integrating binding affinity and eluted ligand data in a combined model demonstrate improved performance for the prediction of MHC-II ligands and T cell epitopes and foreshadow a new generation of improved peptide to MHC-II prediction tools accounting for the plurality of factors that determine natural presentation of antigens. Keywords: MHC-II, Binding predictions, Eluted ligands, T cell epitope, Neural networks, Antigen processing, Machine learning, Mass spectrometry Background MHC-II binding groove, unlike MHC class I, is open at Major histocompatibility complex class II (MHC-II) mole- both ends. -

Antigen Processing and Presentation Pamela Wearsch and Peter Cresswell
Antigen processing and presentation Pamela Wearsch and Peter Cresswell In antigen-presenting cells (APCs), such as dendritic cells (DCs) and B cells, heterogeneous intracellular phagocytosed by APCs, this simple division is not strictly enforced. Indeed, exogenous proteins pathways and mechanisms are responsible for generating complexes of MHC class I and class II internalized by DCs can generate peptide–MHC class I complexes that are recognized by CD8+ T cells, molecules with peptide antigens, and complexes of CD1 molecules with lipid antigens, for presentation a phenomenon referred to as cross-presentation. Similarly, endogenous and viral proteins can generate to T cells. This process — referred to as antigen processing and presentation — allows T cells to peptide–MHC class II complexes that are recognized by CD4+ T cells in a process involving autophagy. continuously assess the intracellular and extracellular milieu for signs of infection or abnormal cell Understanding the processes and mechanisms by which antigens are captured, processed and loaded growth. Although MHC class I molecules typically bind peptides derived from endogenous proteins onto MHC molecules for presentation to T cells provides us with crucial insights that are necessary for and MHC class II molecules typically bind peptides derived from proteins that are endocytosed or the design of vaccines and therapeutic strategies to bolster T-cell responses. The MHC class II pathway The MHC class I pathway MHC class II αβ-chain dimers are assembled in the endoplasmic reticulum (ER) All nucleated cells express MHC class I molecules and present endogenous peptide as a nonameric complex with the invariant chain (Ii), which protects against antigens to CD8+ T cells, but some DC subsets can also present exogenous premature peptide or protein interactions in pre-lysosomal peptides to CD8+ T cells through cross-presentation. -

Antigen Presentation to HLA Class II-Restricted Measles
Proc. Natl. Acad. Sci. USA Vol. 85, pp. 1209-1212, February 1988 Immunology Antigen presentation to HLA class II-restricted measles virus-specific T-cell clones can occur in the absence of the invariant chain (antigen processing/chloroquine/DNA-mediated gene transfer/HLA-DR restriction/major histocompatibility complex) RAFICK P. SEKALY*t, STEVEN JACOBSONt, JOHN R. RICHERT§, CECILE TONNELLE*¶, HENRY F. MCFARLANDt, AND ERIC 0. LONG* *Laboratory of Immunogenetics, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892; tNeuroimmunology Branch, National Institute of Neurological and Communicative Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892; and §Department of Neurology, Georgetown University Medical Center, Washington, DC 20007 Communicated by William E. Paul, November 9, 1987 ABSTRACT A human fibroblast line expressing HLA- The requirements for cell-surface expression of class II a/3 DR1 antigen on its surface was generated by transfection with heterodimers have also been analyzed by DNA-mediated DRa and DRP cDNAs. Expression of the invariant chain gene gene transfer. It was recently shown that the invariant chain was not detectable in the transfected fibroblasts and was not that is transiently associated with a,8 heterodimers intracel- induced by infection with measles virus. Lysis of measles lularly is not required for expression of human MHC class II virus-infected cells occurred with DRM- but not with DR4- antigens at the cell surface (8, 9). These results suggested restricted measles virus-specific cytotoxic T-lymphocyte (CTL) that the invariant chain might be involved in the function of clones and was inhibited by a monoclonal antibody specific for MHC class II antigens, such as binding of processed antigen DR antigen. -

Pa28αβ: the Enigmatic Magic Ring of the Proteasome?
Biomolecules 2014, 4, 566-584; doi:10.3390/biom4020566 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Review PA28: The Enigmatic Magic Ring of the Proteasome? Paolo Cascio Department of Veterinary Sciences, University of Turin, Grugliasco 10095, Italy; E-Mail: [email protected]; Tel.: +39-011-670-9113; Fax: +39-011-670-9138 Received: 4 April 2014; in revised form: 15 May 2014 / Accepted: 8 June 2014 / Published: 19 June 2014 Abstract: PA28 is a -interferon-induced 11S complex that associates with the ends of the 20S proteasome and stimulates in vitro breakdown of small peptide substrates, but not proteins or ubiquitin-conjugated proteins. In cells, PA28 also exists in larger complexes along with the 19S particle, which allows ATP-dependent degradation of proteins; although in vivo a large fraction of PA28 is present as PA28-20S particles whose exact biological functions are largely unknown. Although several lines of evidence strongly indicate that PA28 plays a role in MHC class I antigen presentation, the exact molecular mechanisms of this activity are still poorly understood. Herein, we review current knowledge about the biochemical and biological properties of PA28 and discuss recent findings concerning its role in modifying the spectrum of proteasome’s peptide products, which are important to better understand the molecular mechanisms and biological consequences of PA28 activity. Keywords: PA28; proteasomes; immunoproteasomes; protein degradation; MHC class I antigen presentation; epitopes; antigenic peptides 1. MHC Class I Antigen Presentation The continual presentation of intracellular proteins fragments on major histocompatibility complex (MHC) class I molecules is a process that allows cytotoxic CD8+ T lymphocytes (CTLs) to identify and selectively eliminate cells that synthesize foreign (e.g., viral) or abnormal (e.g., oncogene products) proteins [1,2]. -

Major Histocompatibility Antigens and Antigen-Processing Molecules in Uveal Melanoma1
Vol. 9, 4159–4164, September 15, 2003 Clinical Cancer Research 4159 Major Histocompatibility Antigens and Antigen-processing Molecules in Uveal Melanoma1 Subramanian Krishnakumar, Dhiraj Abhyankar, tigens and the APM was seen in nonepithelioid cell melano- Amirtha Lakshmi Sundaram, mas. There was no correlation with largest tumor diameter. Conclusions: Our data suggest decreased expression of Vaijayanthi Pushparaj, HLA, and APM are seen in uveal melanomas with no ex- Mahesh Palanivelu Shanmugam, and trascleral extension and in nonepithelioid cell melanomas. 2 Jyotirmay Biswas Decreased expression of APM may contribute to decreased Department of Ocular Pathology [S. K., A. L. S., V. P., J. B.] and HLA class I antigen expression. Ocular Oncology [M. P. S.], Medical and Vision Research Foundations, Sankara Nethralaya, Chennai, 600 600, India, and Department of Immunology, Roswell Park Cancer Institute, Buffalo, INTRODUCTION New York 142631 [D. A.] Uveal melanoma is the commonest primary intraocular tumor in Caucasian adults, with an incidence rate of 0.7/100,000. The average survival rate after the diagnosis of ABSTRACT metastatic disease, usually in the liver, is between 2 and 7 Purpose: Malignant transformation of cells is fre- months. No effective treatment for metastatic disease is yet quently associated with abnormalities in the human leuko- available (1). The therapy of uveal melanoma remains problem- cyte antigen (HLA) expression. These abnormalities may atic because of the high rate of metastatic dissemination, irre- play a role in the clinical course of the disease, because HLA spective of the success of treatment of the primary tumor. When antigens mediate interactions of tumor cells with T cells and metastatic disease is diagnosed, patient survival is usually Ͻ1 natural killer cells. -

MHC Structure and Function − Antigen Presentation. Part 2 Estrutura Do MHC E Função − Apresentação De Antígenos
REVIEWING BASIC SCIENCES MHC structure and function − antigen presentation. Part 2 Estrutura do MHC e função − apresentação de antígenos. Parte 2 Anna Carla Goldberg1, Luiz Vicente Rizzo1 ABSTRACT experimental evidence showed that T lymphocytes The second part of this review deals with the molecules and processes recognize portions of antigens (peptides) only when involved in the processing and presentation of the antigenic fragments bound non-covalently to products of the HLA genes. to the T-cell receptor. Though the nature of the antigens presented Crystallographic analysis of the HLAI molecule carried varies, the most significant class of antigens is proteins, processed out in 1987(1) disclosed how these peptides nest in the within the cell to be then recognized in the form of peptides, a external part of the molecule, that is, in the peptide- mechanism that confers an extraordinary degree of precision to this mode of immune response. The efficiency and accuracy of this binding groove. system is also the result of the myriad of mechanisms involved in the processing of proteins and production of peptides, in addition to the capture and recycling of alternative sources aiming to generate PART 2 − PEPTIDE GENERATION: PROCESSING, further diversity in the presentation to T-cells. MECHANISMS OF DIVERSITY, AND ALTERNATIVE SOURCES FOR PRESENTATION Keywords: Major histocompatibility complex; Antigen presentation; Broadly, proteins are degraded inside cells, and peptides HLA genes; Immune response derived through this processing are coupled to HLA molecules and transported to the cell surface. HLAI RESUMO and HLAII molecules transport peptides produced in different cellular compartments (proteasomes and A segunda parte desta revisão trata das moléculas e processos envolvidos no processamento e apresentação dos fragmentos endosomes, respectively), in which different proteolysis antigênicos ao receptor de célula-T. -

Insights Into Cross-Presentation
RESEA r CH HIGHLIGHTS ANTIGEN PRESENTATION pDCs exposed to influenza virus for a short period of time were found to be highly effective stimulators of Prime time: insights into virus-specific CD8+ T-cell prolifera- tion compared with myeloid DCs (mDCs). In contrast to the pathway cross-presentation characterized by Burgdorf et al., cross-presentation by pDCs did not Cross-presentation — the display presentation on MHC class I rely on proteasomal degradation of peptides derived from exogenous molecules are distinct. Based on or cytosolic transit. Instead, viral antigens on MHC class I molecules studies of mouse bone-marrow- antigens were routed directly to recy- — is important for effective derived conventional DCs (cDCs) cling endosomes containing TLR7 immune responses to tumours and presenting the soluble antigen and TLR9, which are crucial for viral infections. Although classical ovalbumin (OVA), it was found that viral recognition. The endosomes of antigen processing pathways are exogenously derived OVA transits pDCs, but not mDCs, acted as large well characterized, the pathways from endosomes into the cytosol and intracellular stores of presynthesized that lead to cross-presentation are is degraded by the proteasome before MHC class I molecules that were less clear. How can a cell physically transport to, and MHC class I load- redistributed to the cell surface early separate endogenous from exter- ing in, the same endosomes. These after exposure to influenza virus. nally derived peptides and select endosomes, identified as essential Therefore, pDCs are unique in their which are presented on MHC class cellular compartments for cross- capacity to rapidly cross-present I molecules? Two studies published presentation but not endogenous viral antigens, owing to endosomal in Nature Immunology have now antigen presentation in cDCs, con- compartments that are specialized for defined two distinct pathways for tained TAP (transporter associated viral recognition and processing.