Viral Interference with Antigen Presentation Jonathan W
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Contrasting Influenza and Respiratory Syncytial Virus
REVIEW published: 02 March 2018 doi: 10.3389/fimmu.2018.00323 Induction and Subversion of Human Protective Immunity: Contrasting influenza and Respiratory Syncytial Virus Stephanie Ascough, Suzanna Paterson and Christopher Chiu* Section of Infectious Diseases and Immunity, Imperial College London, London, United Kingdom Respiratory syncytial virus (RSV) and influenza are among the most important causes of severe respiratory disease worldwide. Despite the clinical need, barriers to devel- oping reliably effective vaccines against these viruses have remained firmly in place for decades. Overcoming these hurdles requires better understanding of human immunity and the strategies by which these pathogens evade it. Although superficially similar, the virology and host response to RSV and influenza are strikingly distinct. Influenza induces robust strain-specific immunity following natural infection, although protection by current vaccines is short-lived. In contrast, even strain-specific protection is incomplete after RSV Edited by: and there are currently no licensed RSV vaccines. Although animal models have been Steven Varga, critical for developing a fundamental understanding of antiviral immunity, extrapolating University of Iowa, United States to human disease has been problematic. It is only with recent translational advances Reviewed by: (such as controlled human infection models and high-dimensional technologies) that the Tara Marlene Strutt, mechanisms responsible for differences in protection against RSV compared to influenza University of Central Florida, have begun to be elucidated in the human context. Influenza infection elicits high-affinity United States Jie Sun, IgA in the respiratory tract and virus-specific IgG, which correlates with protection. Long- Mayo Clinic Minnesota, lived influenza-specific T cells have also been shown to ameliorate disease. -

Antigen Presentation by Dendritic Cells and Their Significance in Antineoplastic Immunotherapy
in vivo 18: 81-100 (2004) Antigen Presentation by Dendritic Cells and their Significance in Antineoplastic Immunotherapy BELA BODEY1,2, STUART E. SIEGEL1,2 and HANS E. KAISER3 1Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA; 2Childrens’ Center for Cancer and Blood Diseases, Childrens’ Hospital Los Angeles, Los Angeles, CA; 3Department of Pathology, School of Medicine, University of Maryland, Baltimore, MD, U.S.A. and Department of General and Experimental Pathology, University of Vienna, Vienna, Austria Abstract. Dendritic cells (DCs) are present in essentially every derived peptides on the surface of major histocompatibility mammalian tissue, where they operate at the interface of innate complex (MHC) molecules and acquire the cellular specialization and acquired immunity by recognizing pathogens and presenting to select and activate naive antigen-specific T lymphocytes. pathogen-derived peptides to T lymphocytes. According to the Immunotherapeutic ideas are based on the ability of the research group of Shortman (1-9), experimental results suggest a mammalian immune system to recognize neoplastically "dual" DC differentiation model, demonstrating the existence of transformed cells. Immunotherapy of human neoplasms has both myeloid-derived (with characteristic IF: CD11b+, CD11c+, always represented a very attractive fourth-modality therapeutic CD8alpha- and DEC205+) and lymphoid-derived DCs (showing approach, especially in light of the many shortcomings of CD11b-, CD11c-, CD8alpha+ and DEC205+ IF). DCs, including conventional surgical, radiation and chemotherapies in the interdigitating cells (IDCs) and Langerhans cells (LCs), are management of neoplastically transformed cells. The cancer characterized by dendritic morphology, high migratory mobility and vaccine approach to therapy is based on the notion that the are the most effective, "professional" cells for antigen presentation immune system could possibly mount a rejection strength response in primary immune responses. -

Antigen Processing (Major Hbitocompatlbility Complex/Class I Moecules/Lymphokines) YOUNG YANG, JAMES B
Proc. Nati. Acad. Sci. USA Vol. 89, pp. 4928-4932, June 1992 Immunology Proteasomes are regulated by interferon y: Implications for antigen processing (major hbitocompatlbility complex/class I moecules/lymphokines) YOUNG YANG, JAMES B. WATERS, KLAUS FROH, AND PER A. PETERSON Department of Immunology, The Scripps Research Institute, La Jolla, CA 92037 Communicated by Frank J. Dixon, February 27, 1992 ABSTRACT Class I major histocompatibility complex strengthened by the findings, presented in this communica- (MHC) molecules present antigenic peptides of cytoplasmic tion, that several proteasomal subunits, including MHC- origin to T cells. As the lengths ofthese peptides seem stried encoded subunits, are regulated by interferon y (IFN--y) and to eight or nine amino acids, an unusual proteolytic system that the incorporation of several more subunits into protea- must play a role in antigen processing. Proteasomes, a major somes appears to depend on the expression of the MHC- extralysosomal proteolytic system, are responsible for the encoded proteasomal subunits. Moreover, the pattern of degradation of cytoplasmic proteins. We demonstrate that expression of IFN-y-regulated subunits suggests complexi- several proteasomal subunits, including MHC-encoded sub- ties in the regulation of proteasomes with respect to its units, are regulated by interferon y. These data and the finding subunit composition, subcellular localization, and its incor- that MHC-encoded and other interferon -regulated protea- poration into larger ubiquitin-related proteolytic complexes. somal subunits are uniquely associated with proteasomes Possible functions for the MHC-encoded and IFN-y- strongly suggest that the immune system has recruited protea- regulated proteasomal subunits in antigen processing are somes for antigen processing. -

Cytoplasmic Proteolysis Antigens on MHC Class II Is Dependent On
Efficient Presentation of Both Cytosolic and Endogenous Transmembrane Protein Antigens on MHC Class II Is Dependent on Cytoplasmic Proteolysis This information is current as of September 28, 2021. Paushali Mukherjee, Aadish Dani, Sumeena Bhatia, Nagendra Singh, Alexander Y. Rudensky, Anna George, Vineeta Bal, Satyajit Mayor and Satyajit Rath J Immunol 2001; 167:2632-2641; ; doi: 10.4049/jimmunol.167.5.2632 Downloaded from http://www.jimmunol.org/content/167/5/2632 References This article cites 51 articles, 20 of which you can access for free at: http://www.jimmunol.org/content/167/5/2632.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 28, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2001 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Efficient Presentation of Both Cytosolic and Endogenous Transmembrane Protein Antigens on MHC Class II Is Dependent on Cytoplasmic Proteolysis1 Paushali Mukherjee,* Aadish Dani,† Sumeena Bhatia,* Nagendra Singh,* Alexander Y. -

UC Irvine UC Irvine Electronic Theses and Dissertations
UC Irvine UC Irvine Electronic Theses and Dissertations Title Computation Models of Virus Dynamics Permalink https://escholarship.org/uc/item/3zb6480f Author Roy, Sarah M. Publication Date 2015 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, IRVINE Computational Models of Virus Dynamics DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Ecology and Evolutionary Biology by Sarah Marie Roy Dissertation Committee: Professor Dominik Wodarz, Chair Associate Professor Robin Bush Associate Professor Kevin Thornton 2015 © 2015 Sarah Marie Roy DEDICATION To my parents and to Hans, in loving memory ii TABLE OF CONTENTS Page LIST OF FIGURES iv ACKNOWLEDGMENTS v CURRICULUM VITAE vi ABSTRACT OF THE DISSERTATION vii INTRODUCTION 1 CHAPTER 1: Infection of HIV-specific CD4 T helper cells and the clonal 13 composition of the response CHAPTER 2: Tissue architecture, feedback regulation, and resilience to 46 viral infection CHAPTER 3: An Agent-Based Model of HIV Coinfection 71 iii LIST OF FIGURES Page Figure 1.1 Outcomes of model (1) assuming a single helper cell cell clone 21 Figure 1.2 Outcomes of model (2) assuming two independently regulated 24 helper cell clones Figure 1.3 Outcomes of model (2) depending on a and b 26 Figure 1.4 Outcomes of model (2) depending on r1 and r2 27 Figure 1.5 Outcomes of model (4) depending on CTL parameters 33 Figure 2.1 Dependence of Sfrac and Dfrac on replication rate, b 53 Figure 2.2 Tissue architecture and resilience to infection according to 55 model (2) Figure 2.3 Uncontrolled growth in the context of negative feedback 59 according to model (2) Figure 2.4 Two different virus persistence equilibria in the stem cell 61 infection model Figure 2.5 Stem cell infection rate vs. -
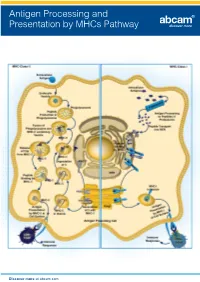
Antigen Processing and Presentation by Mhcs Pathway
Antigen Processing and Presentation by MHCs Pathway Discover more at abcam.com Antigen Processing and Presentation by MHCs Pathway Related antibodies Product This image shows MHC Class II highlights Product Clonality Applications Host Species Reactivity Product code expressing cells in formalin fixed and paraffin embedded human lymph MHC class I antibody [EP1395Y] M Flow Cyt, ICC/IF, IHC-P, IP, WB Rb Hu, Ms, Rat 52922 node, using ab55152. MHC class I antibody [AF6-88.5.5.3] (Biotin) M Flow Cyt Ms Ms 93528 MHC Class II antibody MHC class I antibody [34-1-2S] (FITC) M Flow Cyt Ms Ms 95572 (ab55152) MHC class I antibody [34-1-2S] (Phycoerythrin) M Flow Cyt Ms Ms 95571 MHC Class I alpha antibody [F21-2] M AP, Flow Cyt, IP, WB Ms Chk 23483 Clonality Applications MHC Class 1 H2 Db antibody [27-11-13] - BSA and Azide free M Flow Cyt, FuncS, IHC-Fr Ms Ms 25244 M IHC-P, WB MHC Class 1 H2 Db antibody [28-14-8] (FITC) M Flow Cyt Ms Ms 25056 Host Species cross reactivity MHC Class 1 H2 Db antibody [KH95] M Flow Cyt, FuncS, WB Ms Ms 64373 Ms Hu, RecFrag MHC Class I H2 Dd antibody [34-2-12] M Flow Cyt, FuncS, IHC-Fr, IP Ms Ms 64368 MHC Class I H2 Dk antibody [15-5-5.3] M Flow Cyt Ms Ms 25216 MHC Class II antigens are a valuable tool for studying T helper cell interactions with class II MHC Class I H2 Kb antibody P ELISA, WB Rb Ms 93364 positive antigen presenting cells, as well as the MHC Class I H2 Kd + Dd + q + u + v antibody [1.B.552] (FITC) M Flow Cyt, IP Ms Ms 62228 development of T helper cells since the antigen is present on stromal cells in the thymus. -

Natural Killer Cells Associated with SARS-Cov-2 Viral RNA Shedding
Bao et al. Exp Hematol Oncol (2021) 10:5 https://doi.org/10.1186/s40164-021-00199-1 Experimental Hematology & Oncology LETTER TO THE EDITOR Open Access Natural killer cells associated with SARS-CoV-2 viral RNA shedding, antibody response and mortality in COVID-19 patients Changqian Bao1†, Xiandong Tao2,3†, Wei Cui4†, Yuanyuan Hao1, Shuaike Zheng5, Bin Yi2,3, Tiewen Pan2, Ken H. Young6* and Wenbin Qian1,7* Abstract Coronavirus disease 2019 (COVID-19) is a novel infectious viral disease caused by the severe acute respiratory syn- drome coronavirus 2 (SARS-CoV-2). Two consecutively negative SARS-CoV-2 viral RNA test ( interval 24 hours), improved respiratory symptoms and obvious absorption of infammation in pulmonary imaging are≥ the discharge criteria for COVID-19 patients. The clearance profle of viral RNA in the upper respiratory tract specimens, including nasopharyngeal swab and/or oropharyngeal swabs, is related to innate immune cells such as Natural Killer cells. A total of 168 patients were included for the study. In this cohort, non-severe and severe groups showed signifcant dif- ferences in white blood cells, neutrophils, lymphocytes, basophils and platelets counts, as well as in infection related parameters such as CRP and serum cytokine IL-6. For lymphocyte subsets tests at admission, the severe group dis- played signifcantly lower cell counts than the non-severe group. Higher counts of total T cells, CD4 T cells, CD8 T cells, and NK cells in peripheral blood showed a signifcant correlation with the shorter time taken to+ obtain the frst+ negative viral RNA test and frst positive IgM/ IgG antibody test. -

Targeting Dendritic Cells with Antigen-Delivering Antibodies for Amelioration of Autoimmunity in Animal Models of Multiple Sclerosis and Other Autoimmune Diseases
antibodies Review Targeting Dendritic Cells with Antigen-Delivering Antibodies for Amelioration of Autoimmunity in Animal Models of Multiple Sclerosis and Other Autoimmune Diseases Courtney A. Iberg and Daniel Hawiger * Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, Doisy Research Center, 1205 Carr Lane, St. Louis, MO 63104, USA; [email protected] * Correspondence: [email protected] Received: 31 March 2020; Accepted: 30 April 2020; Published: 15 June 2020 Abstract: The specific targeting of dendritic cells (DCs) using antigen-delivering antibodies has been established to be a highly efficient protocol for the induction of tolerance and protection from autoimmune processes in experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis (MS), as well as in some other animal disease models. As the specific mechanisms of such induced tolerance are being investigated, the newly gained insights may also possibly help to design effective treatments for patients. Here we review approaches applied for the amelioration of autoimmunity in animal models based on antibody-mediated targeting of self-antigens to DCs. Further, we discuss relevant mechanisms of immunological tolerance that underlie such approaches, and we also offer some future perspectives for the application of similar methods in certain related disease settings such as transplantation. Keywords: dendritic cells; tolerance; antigen targeting; chimeric antibodies; autoimmunity; multiple sclerosis; diabetes 1. Introduction Over one hundred years ago, Paul Ehrlich coined the term “horror autotoxicus” to define an immune attack against an organism’s healthy tissues [1]. Since then, our knowledge of the complex mechanisms of the immune system as well as our understanding of the pathogenesis of specific autoimmune diseases have grown tremendously. -

Adaptive Immune Systems
Immunology 101 (for the Non-Immunologist) Abhinav Deol, MD Assistant Professor of Oncology Wayne State University/ Karmanos Cancer Institute, Detroit MI Presentation originally prepared and presented by Stephen Shiao MD, PhD Department of Radiation Oncology Cedars-Sinai Medical Center Disclosures Bristol-Myers Squibb – Contracted Research What is the immune system? A network of proteins, cells, tissues and organs all coordinated for one purpose: to defend one organism from another It is an infinitely adaptable system to combat the complex and endless variety of pathogens it must address Outline Structure of the immune system Anatomy of an immune response Role of the immune system in disease: infection, cancer and autoimmunity Organs of the Immune System Major organs of the immune system 1. Bone marrow – production of immune cells 2. Thymus – education of immune cells 3. Lymph Nodes – where an immune response is produced 4. Spleen – dual role for immune responses (especially antibody production) and cell recycling Origins of the Immune System B-Cell B-Cell Self-Renewing Common Progenitor Natural Killer Lymphoid Cell Progenitor Thymic T-Cell Selection Hematopoetic T-Cell Stem Cell Progenitor Dendritic Cell Myeloid Progenitor Granulocyte/M Macrophage onocyte Progenitor The Immune Response: The Art of War “Know your enemy and know yourself and you can fight a hundred battles without disaster.” -Sun Tzu, The Art of War Immunity: Two Systems and Their Key Players Adaptive Immunity Innate Immunity Dendritic cells (DC) B cells Phagocytes (Macrophages, Neutrophils) Natural Killer (NK) Cells T cells Dendritic Cells: “Commanders-in-Chief” • Function: Serve as the gateway between the innate and adaptive immune systems. -

Dissecting Human Antibody Responses Against Influenza a Viruses and Antigenic Changes That Facilitate Immune Escape
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2018 Dissecting Human Antibody Responses Against Influenza A Viruses And Antigenic Changes That Facilitate Immune Escape Seth J. Zost University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Allergy and Immunology Commons, Immunology and Infectious Disease Commons, Medical Immunology Commons, and the Virology Commons Recommended Citation Zost, Seth J., "Dissecting Human Antibody Responses Against Influenza A Viruses And Antigenic Changes That Facilitate Immune Escape" (2018). Publicly Accessible Penn Dissertations. 3211. https://repository.upenn.edu/edissertations/3211 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3211 For more information, please contact [email protected]. Dissecting Human Antibody Responses Against Influenza A Viruses And Antigenic Changes That Facilitate Immune Escape Abstract Influenza A viruses pose a serious threat to public health, and seasonal circulation of influenza viruses causes substantial morbidity and mortality. Influenza viruses continuously acquire substitutions in the surface glycoproteins hemagglutinin (HA) and neuraminidase (NA). These substitutions prevent the binding of pre-existing antibodies, allowing the virus to escape population immunity in a process known as antigenic drift. Due to antigenic drift, individuals can be repeatedly infected by antigenically distinct influenza strains over the course of their life. Antigenic drift undermines the effectiveness of our seasonal influenza accinesv and our vaccine strains must be updated on an annual basis due to antigenic changes. In order to understand antigenic drift it is essential to know the sites of antibody binding as well as the substitutions that facilitate viral escape from immunity. -

Fitness Costs Limit Influenza a Virus Hemagglutinin Glycosylation
Fitness costs limit influenza A virus hemagglutinin PNAS PLUS glycosylation as an immune evasion strategy Suman R. Dasa,b,1, Scott E. Hensleya,2, Alexandre Davida, Loren Schmidta, James S. Gibbsa, Pere Puigbòc, William L. Incea, Jack R. Benninka, and Jonathan W. Yewdella,3 aLaboratory of Viral Diseases, National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, MD 20892; bEmory Vaccine Center, Emory University, Atlanta, GA 30322; and cNational Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894 AUTHOR SUMMARY Influenza A virus remains an acid substitutions distributed important human pathogen among the four antigenic sites largely because of its ability to recognized by various mono- evade antibodies that neutralize clonal antibodies, indicating viral infectivity. The virus conserved antigenicity among escapes neutralization by alter- the mutants for this mAb (2). ing the target of these anti- Third and highly ironically, bodies, the HA glycoprotein, in escape mutants selected with a process known as antigenic H28-A2 show reduced binding drift. HA attaches the virus to to a remarkably large frac- specific molecules (terminal tion of other monoclonal anti- sialic acid residues) on target bodies (71%) that recognize cells to initiate the infectious one (or a combination) of the cycle. Antibodies that interact four antigenic sites in the glob- with the globular structure ular domain. of the HA protein physically Sequencing of two H28-A2 block virus attachment or the egg-generated escape mutants subsequent HA-mediated (OV1 and OV2) immediately fusion of viral and cellular revealed that their low fre- membranes, thereby neutraliz- quency and high degree of ing viral infectivity. -

What You Need to Know About Innate Immunity
WhatWhat youyou needneed toto knowknow aboutabout innateinnate immunityimmunity JenniferJennifer KoKo MD,MD, PhDPhD ClevelandCleveland ClinicClinic Immunology/PathologyImmunology/Pathology andand LaboratoryLaboratory MedicineMedicine InnateInnate ImmunityImmunity FirstFirst lineline ofof defense,defense, immediateimmediate defensedefense DayDay toto dayday protectionprotection OnlyOnly whenwhen innateinnate defensedefense bypassed,bypassed, evadedevaded oror overwhelmedoverwhelmed isis adaptiveadaptive immunityimmunity requiredrequired NonNon--specificspecific RecognizeRecognize pathogenspathogens inin aa genericgeneric wayway DoesDoes notnot conferconfer longlong lastinglasting oror protectiveprotective immunityimmunity toto hosthost EvolutionarilyEvolutionarily older,older, foundfound inin primitiveprimitive organismsorganisms InnateInnate ImmunityImmunity andand InflammationInflammation 1)1) RespondRespond rapidlyrapidly toto tissuetissue damagedamage physicalphysical andand chemicalchemical barrierbarrier recruitmentrecruitment ofof immuneimmune cellscells toto sitesite ofof injuryinjury 2)2) LimitLimit spreadspread ofof infectioninfection identificationidentification andand removalremoval ofof foreignforeign substancessubstances activationactivation ofof thethe complementcomplement cascadecascade activationactivation ofof coagulationcoagulation cascadecascade 3)3) InitiateInitiate adaptiveadaptive immuneimmune responseresponse antigenantigen presentationpresentation andand cytokinecytokine productionproduction 4)4)