Measurement of Krypton Fission Product Yields from 14 Mev Neutrons on 238U
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
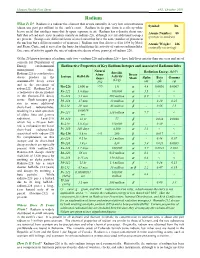
Radium What Is It? Radium Is a Radioactive Element That Occurs Naturally in Very Low Concentrations Symbol: Ra (About One Part Per Trillion) in the Earth’S Crust
Human Health Fact Sheet ANL, October 2001 Radium What Is It? Radium is a radioactive element that occurs naturally in very low concentrations Symbol: Ra (about one part per trillion) in the earth’s crust. Radium in its pure form is a silvery-white heavy metal that oxidizes immediately upon exposure to air. Radium has a density about one- Atomic Number: 88 half that of lead and exists in nature mainly as radium-226, although several additional isotopes (protons in nucleus) are present. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Radium was first discovered in 1898 by Marie Atomic Weight: 226 and Pierre Curie, and it served as the basis for identifying the activity of various radionuclides. (naturally occurring) One curie of activity equals the rate of radioactive decay of one gram (g) of radium-226. Of the 25 known isotopes of radium, only two – radium-226 and radium-228 – have half-lives greater than one year and are of concern for Department of Energy environmental Radioactive Properties of Key Radium Isotopes and Associated Radionuclides management sites. Natural Specific Radiation Energy (MeV) Radium-226 is a radioactive Abun- Decay Isotope Half-Life Activity decay product in the dance Mode Alpha Beta Gamma (Ci/g) uranium-238 decay series (%) (α) (β) (γ) and is the precursor of Ra-226 1,600 yr >99 1.0 α 4.8 0.0036 0.0067 radon-222. Radium-228 is a radioactive decay product Rn-222 3.8 days 160,000 α 5.5 < < in the thorium-232 decay Po-218 3.1 min 290 million α 6.0 < < series. -

The Use of Cosmic-Rays in Detecting Illicit Nuclear Materials
The use of Cosmic-Rays in Detecting Illicit Nuclear Materials Timothy Benjamin Blackwell Department of Physics and Astronomy University of Sheffield This dissertation is submitted for the degree of Doctor of Philosophy 05/05/2015 Declaration I hereby declare that except where specific reference is made to the work of others, the contents of this dissertation are original and have not been submitted in whole or in part for consideration for any other degree or qualification in this, or any other Univer- sity. This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration, except where specifically indicated in the text. Timothy Benjamin Blackwell 05/05/2015 Acknowledgements This thesis could not have been completed without the tremendous support of many people. Firstly I would like to express special appreciation and thanks to my academic supervisor, Dr Vitaly A. Kudryavtsev for his expertise, understanding and encourage- ment. I have enjoyed our many discussions concerning my research topic throughout the PhD journey. I would also like to thank my viva examiners, Professor Lee Thomp- son and Dr Chris Steer, for the time you have taken out of your schedules, so that I may take this next step in my career. Thanks must also be given to Professor Francis Liven, Professor Neil Hyatt and the rest of the Nuclear FiRST DTC team, for the initial oppor- tunity to pursue postgraduate research. Appreciation is also given to the University of Sheffield, the HEP group and EPRSC for providing me with the facilities and funding, during this work. -

Two-Proton Radioactivity 2
Two-proton radioactivity Bertram Blank ‡ and Marek P loszajczak † ‡ Centre d’Etudes Nucl´eaires de Bordeaux-Gradignan - Universit´eBordeaux I - CNRS/IN2P3, Chemin du Solarium, B.P. 120, 33175 Gradignan Cedex, France † Grand Acc´el´erateur National d’Ions Lourds (GANIL), CEA/DSM-CNRS/IN2P3, BP 55027, 14076 Caen Cedex 05, France Abstract. In the first part of this review, experimental results which lead to the discovery of two-proton radioactivity are examined. Beyond two-proton emission from nuclear ground states, we also discuss experimental studies of two-proton emission from excited states populated either by nuclear β decay or by inelastic reactions. In the second part, we review the modern theory of two-proton radioactivity. An outlook to future experimental studies and theoretical developments will conclude this review. PACS numbers: 23.50.+z, 21.10.Tg, 21.60.-n, 24.10.-i Submitted to: Rep. Prog. Phys. Version: 17 December 2013 arXiv:0709.3797v2 [nucl-ex] 23 Apr 2008 Two-proton radioactivity 2 1. Introduction Atomic nuclei are made of two distinct particles, the protons and the neutrons. These nucleons constitute more than 99.95% of the mass of an atom. In order to form a stable atomic nucleus, a subtle equilibrium between the number of protons and neutrons has to be respected. This condition is fulfilled for 259 different combinations of protons and neutrons. These nuclei can be found on Earth. In addition, 26 nuclei form a quasi stable configuration, i.e. they decay with a half-life comparable or longer than the age of the Earth and are therefore still present on Earth. -

"Indoor Radon and Radon Decay Product Measurement Device Protocols"
"Indoor Radon and Radon Decay Product Measurement Device Protocols" U.S. Environmental Protection Agency Office of Air and Radiation (6604J) EPA 402-R-92-004, July 1992 (revised) Table of Contents Disclaimer Acknowledgements Significant Changes in This Revision Radon and Radon Decay Product Measurement Methods Section 1: General Considerations 1.1 Introduction and Background 1.2 General Guidance on Measurement Strategy, Measurement Conditions, Device Location Selection, and Documentation 1.3 Quality Assurance Section 2: Indoor Radon Measurement Device Protocols 2.1 Protocol for Using Continuous Radon Monitors (CR) to Measure Indoor Radon Concentrations 2.2 Protocol for Using Alpha Track Detectors (AT or ATD) to Measure Indoor Radon Concentrations 2.4 Protocol for Using Activated Charcoal Adsorption Devices (AC) to Measure Indoor Radon Concentrations 2.5 Protocol for Using Charcoal Liquid Scintillation (LS) Devices to Measure Indoor Radon Concentrations 2.6 Protocol for Using Grab Radon Sampling (GB, GC, GS), Pump/Collapsible Bag Devices (PB), and Three-Day Integrating Evacuated Scintillation Cells (SC) to Measure Indoor Radon Concentrations 2.7 Interim Protocol for Using Unfiltered Track Detectors (UT) to Measure Indoor Radon Concentrations Section 3: Indoor Radon Decay Product Measurement Device Protocols 3.1 Protocol for Using Continuous Working Level Monitors (CW) to Measure Indoor Radon Decay Product Concentrations 3.2 Protocol for Using Radon Progeny Integrating Sampling Units (RPISU or RP) to Measure Indoor Radon Decay Product Concentrations 3.3 Protocol for Using Grab Sampling-Working Level (GW) to Measure Indoor Radon Decay Product Concentrations Glossary References Please Note: EPA closed its National Radon Proficiency Program (RPP) in 1998. -

Energy What Is a Nuclear Reaction?
4/16/2016 Option C: Energy C.3 : Nuclear Fusion and Fission What is a nuclear reaction? • A nuclear reaction is any reaction that involves the nucleus. • These reactions change the identity of an atom, as opposed to chemical reactions which only involve valence electrons. 1 4/16/2016 The nucleus • The nucleus is made up of protons and neutrons. • We know what the protons do – they provide an electrostatic attraction to the electrons close… but what about the neutrons? The Neutrons • The major function of the neutrons is to hold the nucleus together. • The neutrons provide a strong nuclear force of attraction within the nucleus, counteracting the repulsion between the positively charged protons. 2 4/16/2016 How is the nucleus held together? • In the 1930’s it was first observed that the mass of an atoms nucleus is less than the sum of the masses of the protons + neutrons…? • Some of the mass of the nucleus is converted into energy to hold the nucleus together. 3 4/16/2016 Mass Defect • The difference in mass of the nucleus and it’s parts is referred to as the mass defect, and the energy (e=mc 2) it provided is called the nuclear binding energy. 4 4/16/2016 Nuclear vs. Chemical Reactions • This nuclear binding energy is released during nuclear reactions (fission & fusion), and is ~1,000,000X greater than the chemical bond energy released during chemical reactions. What makes an isotope radioactive? • Elements are radioactive when their nucleus is unstable. • The stabilizing force of the neutrons is effective for smaller elements, though all elements above lead are radioactive. -

Nuclear Data As the Alternative Information of Decay and Analyzer
Usman Kadir, NuCLEAR Data as The alternative Information of decay and Analyzer NuCLEAR Data as The Alternative Information of Decay and Analyzer Usman Kadir Pusat Pendidikan dan Pelatihan BA TAN Abstract NuCLEAR Data is the name of visual, color-coded computer program of "Chart of Nuclides" or "Segre Chart". The program gives you comprehensive nuclear decay data that include data on alpha, beta, gamma, x-ray decay etc., and comprehensive search by energy, intensity, half life, radiation type or name of nuclide. NuCLEAR Data is able to show visual decay product and couldfunction as Multi Nuclides Identification and Multi Elements Identification Analysis. It is constructed using Visual Basic Compiler. Abstak NuCLEAR Data adalah nama program komputer untuk peta nuklida atau Segre Chart; data tervisual dan dikodekan warna. Program ini memberikan anda data peluruhan nuklir secara komprehensif, yang meliputi peluruhan alpha, beta, gamma, sinar-x dan dapat mencari energi, intensitas, waktu paro, jenis radiasi dan nama nuklida. NuCLEAR Data dapat menampilkan produk peluruhan secara visual dan dapat berfungsi sebagai analisis untuk Identifikasi multi nuklida dan identifikasi unsur-unsur. Program ini dibuat dengan Visual Basic. Chart of the Nuclides The "Chart of the Nuclides" or Segre Chart is shown in a colored mode, including details Chart of the Nuclides is a plot of all known for each nuclide's meta-stable states. nuclides as a function of Z (Atomic number), in the y-axis, and N (number of neutrons), in NuCLEAR for Windows 95/98/2000IXP is a the x-axis. visual Nuclear Data Library. It can be used as a standalone information system. -

Modern Physics to Which He Did This Observation, Usually to Within About 0.1%
Chapter 12 NUCLEAR STRUCTURE AND RADIOACTIVITY Radioactive isotopes have proven to be valuable tools for medical diagnosis. The photo shows gamma-ray emission from a man who has been treated with a radioactive element. The radioactivity concentrates in locations where there are active cancer tumors, which show as bright areas in the gamma-ray scan. This patient’s cancer has spread from his prostate gland to several other locations in his body. 370 Chapter 12 | Nuclear Structure and Radioactivity The nucleus lies at the center of the atom, occupying only 10−15 of its volume but providing the electrical force that holds the atom together. Within the nucleus there are Z positive charges. To keep these charges from flying apart, the nuclear force must supply an attraction that overcomes their electrical repulsion. This nuclear force is the strongest of the known forces; it provides nuclear binding energies that are millions of times stronger than atomic binding energies. There are many similarities between atomic structure and nuclear structure, which will make our study of the properties of the nucleus somewhat easier. Nuclei are subject to the laws of quantum physics. They have ground and excited states and emit photons in transitions between the excited states. Just like atomic states, nuclear states can be labeled by their angular momentum. There are, however, two major differences between the study of atomic and nuclear properties. In atomic physics, the electrons experience the force provided by an external agent, the nucleus; in nuclear physics, there is no such external agent. In contrast to atomic physics, in which we can often consider the interactions among the electrons as a perturbation to the primary interaction between electrons and nucleus, in nuclear physics the mutual interaction of the nuclear constituents is just what provides the nuclear force, so we cannot treat this complicated many- body problem as a correction to a single-body problem. -

What Is Radioactivity?
General Academic Unit Radon Alert INVESTIGATION 2 WHAT IS RADIOACTIVITY? INTRODUCTION The study of probability allows us to make predictions about the occurrence of certain events such as birth, death, and accident rates, and in this case, the breakdown of atomic nuclei during the radioactive decay process. Radioactivity is emitted when particles or energy rays are given off by an atom's nucleus (See Figure 1). Three different kinds of radiation can be given off: alpha, beta, or gamma (Figure 2). During this “decay” process, the element spontaneously gives off particles or energy rays, known as radiation. Radon-222 is the direct product of the decay of the most common radium isotope, radium-226. Radium is a product, several steps removed, of the decay of the most abundant uranium isotope, uranium-238. Radon-222 breaks down into its decay product isotopes, polonium-218, among others. Decay product isotopes are sometimes referred to as progeny or daughters. Each element has a characteristic average rate of decay that doesn't change. The time it takes for one-half of the atoms in a given radioactive sample to decay is called the half-life of that isotope. Each radioactive isotope has its own unique half-life, which is totally unaffected by temperature, pressure, and other factors. In this exercise you will conduct an investigation that simulates radioactive half-life and its relationship to statistical probability. OBJECTIVE To apply the process of probability to the concepts of radioactive decay and radioac- tive half-life. MATERIALS • One large bag of m&m’s (plain) Shells Through Which Electrons Travel Around the Nucleus Nucleus Protons Neutrons Figure 1. -

Synthesis and Spectroscopic Properties of Transfermium Isotopes with Z = 105, 106 and 107
FACULTY OF MATHEMATICS, PHYSICS AND INFORMATICS COMENIUS UNIVERSITY BRATISLAVA Department of Nuclear Physics Synthesis and spectroscopic properties of transfermium isotopes with Z = 105, 106 and 107 Accademic Dissertation for the Degree of Doctor of Philosophy by Branislav Streicherˇ Supervisors: Bratislava 2006 Prof. RNDr. Stefanˇ S´aro,ˇ DrSc. Dr. F.P. Heßberger Abstract The quest for production of new elements has been on for several decades. On the way up the ladder of nuclear chart the systematic research of nuclear properties of elements in transfermium region has been severely overlooked. This drawback is being rectified in past few years by systematic synthesis of especially even-even and odd-A isotopes of these elements. This work proceeds forward also with major contribution of velocity filter SHIP, placed at GSI, Darmstadt. This experimental device represents a unique possibility due to high (up to 1pµA) beam currents provided by UNILAC accelerator and advancing detection systems to study by means of decay α-γ spectroscopy the nuclear structure of isotopes for the elements, possibly up to proton number Z = 110. As the low lying single-particle levels are especially determined by the unpaired nucleon, the odd mass nuclei provide a valuable source of information about the nuclear structure. Such results can be directly compared with the predictions of the calculations based on macroscopic-microscopic model of nuclear matter, thus proving an unambiguous test of the correctness of present models and their power to predict nuclear properties towards yet unknown regions. This work concentrates on the spectroscopic analysis of few of such nuclei. Namely it deals with isotopes 261Sg and 257Rf with one unpaired neutron, as well as isotopes 257Db and 253Lr with one unpaired proton configuration. -

The Supply of Medical Radioisotopes
Nuclear Development November 2010 The Supply of Medical Radioisotopes Review of Potential Molybdenum-99/ Technetium-99m Production Technologies NUCLEAR ENERGY AGENCY The Supply of Medical Radioisotopes: Review of Potential Molybdenum-99/Technetium-99m Production Technologies © OECD 2010 NUCLEAR ENERGY AGENCY ORGANISATION FOR ECONOMIC CO-OPERATION AND DEVELOPMENT 2 1 FOREWORD At the request of its member countries, the OECD Nuclear Energy Agency (NEA) has become involved in global efforts to ensure a reliable supply of molybdenum-99 (99Mo) and its decay product, technetium-99m (99mTc), the most widely used medical radioisotope. The NEA Steering Committee for Nuclear Energy established the High-level Group on the Security of Supply of Medical Radioisotopes (HLG-MR) in April 2009. The main objective of the HLG-MR is to strengthen the reliability of 99Mo and 99mTc supply in the short, medium and long term. In order to reach this objective, the group has been reviewing the 99Mo supply chain, working to identify the key areas of vulnerability, the issues that need to be addressed and the mechanisms that could be used to help resolve them. A report on the economic study of the 99Mo supply chain was issued in September 2010. Among the actions requested by the HLG-MR was a review of other potential methods of producing 99Mo, of which the two main methods are reactor-based and accelerator-based. The HLG- MR requested both the development of a set of criteria for assessing the existing and proposed methods, as well as an analysis, where possible, of those methods using these criteria. This report provides a set of criteria and an initial assessment of the situation at the present time, based mainly on published sources of information. -

Radioactivity
AccessScience from McGraw-Hill Education Page 1 of 43 www.accessscience.com Radioactivity Contributed by: Joseph H. Hamilton Publication year: 2014 A phenomenon resulting from an instability of the atomic nucleus in certain atoms whereby the nucleus experiences a spontaneous but measurably delayed nuclear transition or transformation with the resulting emission of radiation. The discovery of radioactivity by Henri Becquerel in 1896 was an indirect consequence of the discovery of x-rays a few months earlier by Wilhelm Roentgen, and marked the birth of nuclear physics. See also: X-RAYS. On the other hand, nuclear physics can also be said to begin with the proposal by Ernest Rutherford in 1911 that atoms have a nucleus. On the basis of the scattering of alpha particles (emitted in radioactive decay) by gold foils, Rutherford proposed a solar model of atoms, where negatively charged electrons orbit the tiny nucleus, which contains all the positive charge and essentially all the mass of the atom, as planets orbit around the Sun. The attractive Coulomb electrical force holds the electrons in orbit about the nucleus. Atoms have radii of about 10,−10 m and the nuclei of atoms have radii about 2 × 10,−15 m, so atoms are mostly empty space, like the solar system. Niels Bohr proposed a theoretical model for the atom that removed certain difficulties of the Rutherford model. See also: ATOMIC STRUCTURE AND SPECTRA. However, it was only after the discovery of the neutron in 1932 that a proper understanding was achieved of the particles that compose the nucleus of the atom. -

What Is Radon? • • •
Earth Science Unit Radon Alert TEACHER'S NOTES 3 WHAT IS RADON? BACKGROUND Students may have some difficulty understanding the relationship among the isotopes in the uranium-238 decay series (Figure 1). Each time an alpha particle is emitted during the series, the number of protons decreases by 2 and the number of neutrons decreases by 2. This is because an alpha particle consists of 2 protons and 2 neutrons. The atomic number is the number of protons. The mass number (i.e., the 238 in uranium-238) is the number of protons plus the number of neutrons. When a beta particle is emitted, the mass number stays the same because no protons or neutrons are emitted. A beta particle is an electron, with very slight mass and a negative charge, and is formed when a neutron breaks apart into a proton and electron. The atomic number increases by one when a beta particle is emitted. This can be thought of as a conversion of one neutron into one proton to compensate for the loss of the negatively-charged beta particle. Thus, even though the atomic number changes, the mass number stays the same during beta emission. The half-life of each isotope is given below its symbol in the figure. Half-life is an important characteristic of each isotope, and can be a difficult one for students to grasp (see Investigation 2). MINIMUM RECOMMENDED TIME ALLOCATION One class period. STUDENT RESPONSES Question 3: Radon would be a lesser health threat if the half-life was either very short (it would not make its way out of the soil before being trans- formed from a gas to a solid) or very long (it would escape from the house before it decayed to polonium).