Manual for Reactor Produced Radioisotopes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Safeguards at Research Reactors: Current Practices, Future Directions
FEATURES Safeguards at research reactors: Current practices, future directions Some new verification measures are being introduced to improve the efficiency and the effectiveness of the Agency's safeguards by Giancarlo /approximately 180 research reactors and crit- immersed in a large pool of water that provides Zuccaro-Labellarte ical assemblies currently are under IAEA safe- both cooling and neutron moderation. The fuel and guards. The vast majority of the research reac- assemblies in the core of a swimming pool reac- Robert Fagerholm tors operate at relatively low power levels (10 tor are normally visible and accessible for safe- megawatts-thermal or lower) and the critical guards measurements. assemblies at virtually zero power.* From a Other types of research reactors operate at safeguards standpoint, this is important since a higher power levels (exceeding 10 megawatts- reactor's power level is a determining factor of thermal). They need more powerful heat its capability to produce plutonium. Along with removal systems and are therefore normally high-enriched uranium (HEU) and uranium- enclosed in core vessels and equipped with 233, plutonium is considered a "direct use" coolant pumps and heat exchangers. The fuel material which could be diverted for the pro- elements in the reactor core at these installa- duction of nuclear weapons. tions are usually not visible or accessible for In this article, the IAEA's safeguards safeguards measurements. implementation at research reactors is Research reactors are widely used for scien- addressed, including aspects related to diver- tific investigations and various applications. sion and clandestine production scenarios and Neutrons produced by research reactors provide main verification activities. -
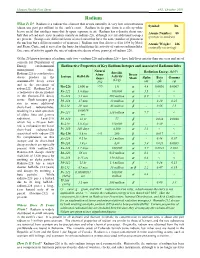
Radium What Is It? Radium Is a Radioactive Element That Occurs Naturally in Very Low Concentrations Symbol: Ra (About One Part Per Trillion) in the Earth’S Crust
Human Health Fact Sheet ANL, October 2001 Radium What Is It? Radium is a radioactive element that occurs naturally in very low concentrations Symbol: Ra (about one part per trillion) in the earth’s crust. Radium in its pure form is a silvery-white heavy metal that oxidizes immediately upon exposure to air. Radium has a density about one- Atomic Number: 88 half that of lead and exists in nature mainly as radium-226, although several additional isotopes (protons in nucleus) are present. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Radium was first discovered in 1898 by Marie Atomic Weight: 226 and Pierre Curie, and it served as the basis for identifying the activity of various radionuclides. (naturally occurring) One curie of activity equals the rate of radioactive decay of one gram (g) of radium-226. Of the 25 known isotopes of radium, only two – radium-226 and radium-228 – have half-lives greater than one year and are of concern for Department of Energy environmental Radioactive Properties of Key Radium Isotopes and Associated Radionuclides management sites. Natural Specific Radiation Energy (MeV) Radium-226 is a radioactive Abun- Decay Isotope Half-Life Activity decay product in the dance Mode Alpha Beta Gamma (Ci/g) uranium-238 decay series (%) (α) (β) (γ) and is the precursor of Ra-226 1,600 yr >99 1.0 α 4.8 0.0036 0.0067 radon-222. Radium-228 is a radioactive decay product Rn-222 3.8 days 160,000 α 5.5 < < in the thorium-232 decay Po-218 3.1 min 290 million α 6.0 < < series. -

1 11. Nuclear Chemistry 11.1 Stable and Unstable Nuclides Very Large
11. Nuclear Chemistry Chemical reactions occur as a result of loosing/gaining and sharing electrons in the valance shell which is far away from the atomic nucleus as we described in previous chapters in chemical bonding. In chemical reactions identity of the elements (atomic) and the makeup of the nuclei (mass due to protons and neutrons) is preserved which is reflected in the Law of Conservation of mass. This idea of atomic nucleus is always stable was shattered as Henri Becquerel discovered radioactivity in uranium compound where uranium nuclei changes or undergo nuclear reactions where nuclei of an element is transformed into nuclei of different element(s) while emitting ionization radiation. Marie Curie also began a study of radioactivity in a different form of uranium ore called pitchblende and she discovered the existence of two more highly radioactive new elements radium and polonium formed as the products during the decay of unstable nuclide of uranium-235. Curie measure that the radiation emanated was proportional to the amount (moles or number of nuclides) of radioactive element present, and she proposed that radiation was a property nucleus of an unstable atom. The area of chemistry that focuses on the nuclear changes is called nuclear chemistry. What changes in a nuclide result from the loss of each of the following? a) An alpha particle. b) A gamma ray. c) An electron. d) A neutron. e) A proton. Answer: a), c), d), e) 11.1 Stable and Unstable Nuclides There are stable and unstable radioactive nuclides. Unstable nuclides emit subatomic particles, with alpha −α, beta −β, gamma −γ, proton-p, neutrons-n being the most common. -

The Use of Cosmic-Rays in Detecting Illicit Nuclear Materials
The use of Cosmic-Rays in Detecting Illicit Nuclear Materials Timothy Benjamin Blackwell Department of Physics and Astronomy University of Sheffield This dissertation is submitted for the degree of Doctor of Philosophy 05/05/2015 Declaration I hereby declare that except where specific reference is made to the work of others, the contents of this dissertation are original and have not been submitted in whole or in part for consideration for any other degree or qualification in this, or any other Univer- sity. This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration, except where specifically indicated in the text. Timothy Benjamin Blackwell 05/05/2015 Acknowledgements This thesis could not have been completed without the tremendous support of many people. Firstly I would like to express special appreciation and thanks to my academic supervisor, Dr Vitaly A. Kudryavtsev for his expertise, understanding and encourage- ment. I have enjoyed our many discussions concerning my research topic throughout the PhD journey. I would also like to thank my viva examiners, Professor Lee Thomp- son and Dr Chris Steer, for the time you have taken out of your schedules, so that I may take this next step in my career. Thanks must also be given to Professor Francis Liven, Professor Neil Hyatt and the rest of the Nuclear FiRST DTC team, for the initial oppor- tunity to pursue postgraduate research. Appreciation is also given to the University of Sheffield, the HEP group and EPRSC for providing me with the facilities and funding, during this work. -

Two-Proton Radioactivity 2
Two-proton radioactivity Bertram Blank ‡ and Marek P loszajczak † ‡ Centre d’Etudes Nucl´eaires de Bordeaux-Gradignan - Universit´eBordeaux I - CNRS/IN2P3, Chemin du Solarium, B.P. 120, 33175 Gradignan Cedex, France † Grand Acc´el´erateur National d’Ions Lourds (GANIL), CEA/DSM-CNRS/IN2P3, BP 55027, 14076 Caen Cedex 05, France Abstract. In the first part of this review, experimental results which lead to the discovery of two-proton radioactivity are examined. Beyond two-proton emission from nuclear ground states, we also discuss experimental studies of two-proton emission from excited states populated either by nuclear β decay or by inelastic reactions. In the second part, we review the modern theory of two-proton radioactivity. An outlook to future experimental studies and theoretical developments will conclude this review. PACS numbers: 23.50.+z, 21.10.Tg, 21.60.-n, 24.10.-i Submitted to: Rep. Prog. Phys. Version: 17 December 2013 arXiv:0709.3797v2 [nucl-ex] 23 Apr 2008 Two-proton radioactivity 2 1. Introduction Atomic nuclei are made of two distinct particles, the protons and the neutrons. These nucleons constitute more than 99.95% of the mass of an atom. In order to form a stable atomic nucleus, a subtle equilibrium between the number of protons and neutrons has to be respected. This condition is fulfilled for 259 different combinations of protons and neutrons. These nuclei can be found on Earth. In addition, 26 nuclei form a quasi stable configuration, i.e. they decay with a half-life comparable or longer than the age of the Earth and are therefore still present on Earth. -

"Indoor Radon and Radon Decay Product Measurement Device Protocols"
"Indoor Radon and Radon Decay Product Measurement Device Protocols" U.S. Environmental Protection Agency Office of Air and Radiation (6604J) EPA 402-R-92-004, July 1992 (revised) Table of Contents Disclaimer Acknowledgements Significant Changes in This Revision Radon and Radon Decay Product Measurement Methods Section 1: General Considerations 1.1 Introduction and Background 1.2 General Guidance on Measurement Strategy, Measurement Conditions, Device Location Selection, and Documentation 1.3 Quality Assurance Section 2: Indoor Radon Measurement Device Protocols 2.1 Protocol for Using Continuous Radon Monitors (CR) to Measure Indoor Radon Concentrations 2.2 Protocol for Using Alpha Track Detectors (AT or ATD) to Measure Indoor Radon Concentrations 2.4 Protocol for Using Activated Charcoal Adsorption Devices (AC) to Measure Indoor Radon Concentrations 2.5 Protocol for Using Charcoal Liquid Scintillation (LS) Devices to Measure Indoor Radon Concentrations 2.6 Protocol for Using Grab Radon Sampling (GB, GC, GS), Pump/Collapsible Bag Devices (PB), and Three-Day Integrating Evacuated Scintillation Cells (SC) to Measure Indoor Radon Concentrations 2.7 Interim Protocol for Using Unfiltered Track Detectors (UT) to Measure Indoor Radon Concentrations Section 3: Indoor Radon Decay Product Measurement Device Protocols 3.1 Protocol for Using Continuous Working Level Monitors (CW) to Measure Indoor Radon Decay Product Concentrations 3.2 Protocol for Using Radon Progeny Integrating Sampling Units (RPISU or RP) to Measure Indoor Radon Decay Product Concentrations 3.3 Protocol for Using Grab Sampling-Working Level (GW) to Measure Indoor Radon Decay Product Concentrations Glossary References Please Note: EPA closed its National Radon Proficiency Program (RPP) in 1998. -

Background and Derivation of ANS-5.4 Standard Fission Product Release Model
NUREG/CR-7003 PNNL-18490 Background and Derivation of ANS-5.4 Standard Fission Product Release Model Office of Nuclear Regulatory Research AVAILABILITY OF REFERENCE MATERIALS IN NRC PUBLICATIONS NRC Reference Material Non-NRC Reference Material As of November 1999, you may electronically access Documents available from public and special technical NUREG-series publications and other NRC records at libraries include all open literature items, such as NRC’s Public Electronic Reading Room at books, journal articles, and transactions, Federal http://www.nrc.gov/reading-rm.html. Register notices, Federal and State legislation, and Publicly released records include, to name a few, congressional reports. Such documents as theses, NUREG-series publications; Federal Register notices; dissertations, foreign reports and translations, and applicant, licensee, and vendor documents and non-NRC conference proceedings may be purchased correspondence; NRC correspondence and internal from their sponsoring organization. memoranda; bulletins and information notices; inspection and investigative reports; licensee event reports; and Commission papers and their attachments. Copies of industry codes and standards used in a substantive manner in the NRC regulatory process are NRC publications in the NUREG series, NRC maintained at— regulations, and Title 10, Energy, in the Code of The NRC Technical Library Federal Regulations may also be purchased from one Two White Flint North of these two sources. 11545 Rockville Pike 1. The Superintendent of Documents Rockville, MD 20852–2738 U.S. Government Printing Office Mail Stop SSOP Washington, DC 20402–0001 These standards are available in the library for Internet: bookstore.gpo.gov reference use by the public. Codes and standards are Telephone: 202-512-1800 usually copyrighted and may be purchased from the Fax: 202-512-2250 originating organization or, if they are American 2. -

Energy What Is a Nuclear Reaction?
4/16/2016 Option C: Energy C.3 : Nuclear Fusion and Fission What is a nuclear reaction? • A nuclear reaction is any reaction that involves the nucleus. • These reactions change the identity of an atom, as opposed to chemical reactions which only involve valence electrons. 1 4/16/2016 The nucleus • The nucleus is made up of protons and neutrons. • We know what the protons do – they provide an electrostatic attraction to the electrons close… but what about the neutrons? The Neutrons • The major function of the neutrons is to hold the nucleus together. • The neutrons provide a strong nuclear force of attraction within the nucleus, counteracting the repulsion between the positively charged protons. 2 4/16/2016 How is the nucleus held together? • In the 1930’s it was first observed that the mass of an atoms nucleus is less than the sum of the masses of the protons + neutrons…? • Some of the mass of the nucleus is converted into energy to hold the nucleus together. 3 4/16/2016 Mass Defect • The difference in mass of the nucleus and it’s parts is referred to as the mass defect, and the energy (e=mc 2) it provided is called the nuclear binding energy. 4 4/16/2016 Nuclear vs. Chemical Reactions • This nuclear binding energy is released during nuclear reactions (fission & fusion), and is ~1,000,000X greater than the chemical bond energy released during chemical reactions. What makes an isotope radioactive? • Elements are radioactive when their nucleus is unstable. • The stabilizing force of the neutrons is effective for smaller elements, though all elements above lead are radioactive. -

The Delimiting/Frontier Lines of the Constituents of Matter∗
The delimiting/frontier lines of the constituents of matter∗ Diógenes Galetti1 and Salomon S. Mizrahi2 1Instituto de Física Teórica, Universidade Estadual Paulista (UNESP), São Paulo, SP, Brasily 2Departamento de Física, CCET, Universidade Federal de São Carlos, São Carlos, SP, Brasilz (Dated: October 23, 2019) Abstract Looking at the chart of nuclides displayed at the URL of the International Atomic Energy Agency (IAEA) [1] – that contains all the known nuclides, the natural and those produced artificially in labs – one verifies the existence of two, not quite regular, delimiting lines between which dwell all the nuclides constituting matter. These lines are established by the highly unstable radionuclides located the most far away from those in the central locus, the valley of stability. Here, making use of the “old” semi-empirical mass formula for stable nuclides together with the energy-time uncertainty relation of quantum mechanics, by a simple calculation we show that the obtained frontier lines, for proton and neutron excesses, present an appreciable agreement with the delimiting lines. For the sake of presenting a somewhat comprehensive panorama of the matter in our Universe and their relation with the frontier lines, we narrate, in brief, what is currently known about the astrophysical nucleogenesis processes. Keywords: chart of nuclides, nucleogenesis, nuclide mass formula, valley/line of stability, energy-time un- certainty relation, nuclide delimiting/frontier lines, drip lines arXiv:1909.07223v2 [nucl-th] 22 Oct 2019 ∗ This manuscript is based on an article that was published in Brazilian portuguese language in the journal Revista Brasileira de Ensino de Física, 2018, 41, e20180160. http://dx.doi.org/10.1590/ 1806-9126-rbef-2018-0160. -

Nuclear Data As the Alternative Information of Decay and Analyzer
Usman Kadir, NuCLEAR Data as The alternative Information of decay and Analyzer NuCLEAR Data as The Alternative Information of Decay and Analyzer Usman Kadir Pusat Pendidikan dan Pelatihan BA TAN Abstract NuCLEAR Data is the name of visual, color-coded computer program of "Chart of Nuclides" or "Segre Chart". The program gives you comprehensive nuclear decay data that include data on alpha, beta, gamma, x-ray decay etc., and comprehensive search by energy, intensity, half life, radiation type or name of nuclide. NuCLEAR Data is able to show visual decay product and couldfunction as Multi Nuclides Identification and Multi Elements Identification Analysis. It is constructed using Visual Basic Compiler. Abstak NuCLEAR Data adalah nama program komputer untuk peta nuklida atau Segre Chart; data tervisual dan dikodekan warna. Program ini memberikan anda data peluruhan nuklir secara komprehensif, yang meliputi peluruhan alpha, beta, gamma, sinar-x dan dapat mencari energi, intensitas, waktu paro, jenis radiasi dan nama nuklida. NuCLEAR Data dapat menampilkan produk peluruhan secara visual dan dapat berfungsi sebagai analisis untuk Identifikasi multi nuklida dan identifikasi unsur-unsur. Program ini dibuat dengan Visual Basic. Chart of the Nuclides The "Chart of the Nuclides" or Segre Chart is shown in a colored mode, including details Chart of the Nuclides is a plot of all known for each nuclide's meta-stable states. nuclides as a function of Z (Atomic number), in the y-axis, and N (number of neutrons), in NuCLEAR for Windows 95/98/2000IXP is a the x-axis. visual Nuclear Data Library. It can be used as a standalone information system. -

Modern Physics to Which He Did This Observation, Usually to Within About 0.1%
Chapter 12 NUCLEAR STRUCTURE AND RADIOACTIVITY Radioactive isotopes have proven to be valuable tools for medical diagnosis. The photo shows gamma-ray emission from a man who has been treated with a radioactive element. The radioactivity concentrates in locations where there are active cancer tumors, which show as bright areas in the gamma-ray scan. This patient’s cancer has spread from his prostate gland to several other locations in his body. 370 Chapter 12 | Nuclear Structure and Radioactivity The nucleus lies at the center of the atom, occupying only 10−15 of its volume but providing the electrical force that holds the atom together. Within the nucleus there are Z positive charges. To keep these charges from flying apart, the nuclear force must supply an attraction that overcomes their electrical repulsion. This nuclear force is the strongest of the known forces; it provides nuclear binding energies that are millions of times stronger than atomic binding energies. There are many similarities between atomic structure and nuclear structure, which will make our study of the properties of the nucleus somewhat easier. Nuclei are subject to the laws of quantum physics. They have ground and excited states and emit photons in transitions between the excited states. Just like atomic states, nuclear states can be labeled by their angular momentum. There are, however, two major differences between the study of atomic and nuclear properties. In atomic physics, the electrons experience the force provided by an external agent, the nucleus; in nuclear physics, there is no such external agent. In contrast to atomic physics, in which we can often consider the interactions among the electrons as a perturbation to the primary interaction between electrons and nucleus, in nuclear physics the mutual interaction of the nuclear constituents is just what provides the nuclear force, so we cannot treat this complicated many- body problem as a correction to a single-body problem. -

18 IGORR Conference IAEA Workshop on Safety Reassessment
18th IGORR Conference and IAEA Workshop 18th IGORR Conference and IAEA Workshop on Safety Reassessment of Research Reactors in Light of the Lessons Learned from the Fukushima Daiichi Accident, J7-TR-54790 International Conference Centre Sydney Darling Harbour, Sydney, Australia Sunday 3 – Thursday 7 December 2017 Sunday 3 December 17:00 Registration Reception Drinks and canapés Register for the Conference 18th IGORR Conference and IAEA Workshop Monday 4 December 08:20 Opening Session (Room C4.1) Chair: ANSTO Welcome to Country Welcome from ANSTO, IGORR & IAEA 09:00 General Session (Room C4.1) Chairs: David Vittorio & Gilles Bignan Andrea Borio di Tigliole: IAEA activities to support sustainable operation of and access to research reactors Gilles Bignan: The CEA scientific and technical offer as a designated ICERR by the IAEA: First feedback with the prime Affiliates Alexander Tuzov: RIAR as IAEA ICERR: Pilot technical cooperation projects and future prospects Sean O’Kelly: The first 50 years of operation of the ATR at the Idaho National Laboratory Khalid Almarri: A qualitative study for establishing the conditions for the successful implementation of public private partnerships in research reactor project in newcomer contries 10:40 Morning Tea Break (Room C4.4) 11:00 IAEA Workshop (Room C4.1) Chair: David Sears David Sears: IAEA Activities on the safety of Research Reactors Alexander Sapozhnikov: New safety requirements addressing feedback from the Fukushima Daiichi accident Mark Summerfield: Some thoughts on operator intervention arising