Legius Syndrome Genetic Testing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Piggybac Mutagenesis and Exome Sequencing Identify Genetic Driver
Noorani et al. Genome Biology (2020) 21:181 https://doi.org/10.1186/s13059-020-02092-2 RESEARCH Open Access PiggyBac mutagenesis and exome sequencing identify genetic driver landscapes and potential therapeutic targets of EGFR-mutant gliomas Imran Noorani1,2* , Jorge de la Rosa1†, Yoon Ha Choi1,3†, Alexander Strong1, Hannes Ponstingl1, M. S. Vijayabaskar1, Jusung Lee3, Eunmin Lee3, Angela Richard-Londt4, Mathias Friedrich1,5, Federica Furlanetto5, Rocio Fuente1, Ruby Banerjee1, Fengtang Yang1, Frances Law1, Colin Watts2,6, Roland Rad5, George Vassiliou1, Jong Kyoung Kim3, Thomas Santarius2, Sebastian Brandner4 and Allan Bradley1* * Correspondence: [email protected]; [email protected] Abstract †Jorge de la Rosa and Yoonha Choi contributed equally to this work. Background: Glioma is the most common intrinsic brain tumor and also occurs in 1The Wellcome Trust Sanger the spinal cord. Activating EGFR mutations are common in IDH1 wild-type gliomas. Institute, Wellcome Trust Genome However, the cooperative partners of EGFR driving gliomagenesis remain poorly Campus, Hinxton, Cambridgeshire CB10 1SA, UK understood. Full list of author information is Results: We explore EGFR-mutant glioma evolution in conditional mutant mice by available at the end of the article whole-exome sequencing, transposon mutagenesis forward genetic screening, and transcriptomics. We show mutant EGFR is sufficient to initiate gliomagenesis in vivo, both in the brain and spinal cord. We identify significantly recurrent somatic alterations in these gliomas including mutant EGFR amplifications and Sub1, Trp53, and Tead2 loss-of-function mutations. Comprehensive functional characterization of 96 gliomas by genome-wide piggyBac insertional mutagenesis in vivo identifies 281 known and novel EGFR-cooperating driver genes, including Cdkn2a, Nf1, Spred1, and Nav3. -

Megalencephaly and Macrocephaly
277 Megalencephaly and Macrocephaly KellenD.Winden,MD,PhD1 Christopher J. Yuskaitis, MD, PhD1 Annapurna Poduri, MD, MPH2 1 Department of Neurology, Boston Children’s Hospital, Boston, Address for correspondence Annapurna Poduri, Epilepsy Genetics Massachusetts Program, Division of Epilepsy and Clinical Electrophysiology, 2 Epilepsy Genetics Program, Division of Epilepsy and Clinical Department of Neurology, Fegan 9, Boston Children’s Hospital, 300 Electrophysiology, Department of Neurology, Boston Children’s Longwood Avenue, Boston, MA 02115 Hospital, Boston, Massachusetts (e-mail: [email protected]). Semin Neurol 2015;35:277–287. Abstract Megalencephaly is a developmental disorder characterized by brain overgrowth secondary to increased size and/or numbers of neurons and glia. These disorders can be divided into metabolic and developmental categories based on their molecular etiologies. Metabolic megalencephalies are mostly caused by genetic defects in cellular metabolism, whereas developmental megalencephalies have recently been shown to be caused by alterations in signaling pathways that regulate neuronal replication, growth, and migration. These disorders often lead to epilepsy, developmental disabilities, and Keywords behavioral problems; specific disorders have associations with overgrowth or abnor- ► megalencephaly malities in other tissues. The molecular underpinnings of many of these disorders are ► hemimegalencephaly now understood, providing insight into how dysregulation of critical pathways leads to ► -

Cardiomyopathy Precision Panel Overview Indications
Cardiomyopathy Precision Panel Overview Cardiomyopathies are a group of conditions with a strong genetic background that structurally hinder the heart to pump out blood to the rest of the body due to weakness in the heart muscles. These diseases affect individuals of all ages and can lead to heart failure and sudden cardiac death. If there is a family history of cardiomyopathy it is strongly recommended to undergo genetic testing to be aware of the family risk, personal risk, and treatment options. Most types of cardiomyopathies are inherited in a dominant manner, which means that one altered copy of the gene is enough for the disease to present in an individual. The symptoms of cardiomyopathy are variable, and these diseases can present in different ways. There are 5 types of cardiomyopathies, the most common being hypertrophic cardiomyopathy: 1. Hypertrophic cardiomyopathy (HCM) 2. Dilated cardiomyopathy (DCM) 3. Restrictive cardiomyopathy (RCM) 4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) 5. Isolated Left Ventricular Non-Compaction Cardiomyopathy (LVNC). The Igenomix Cardiomyopathy Precision Panel serves as a diagnostic and tool ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes. Indications The Igenomix Cardiomyopathy Precision Panel is indicated in those cases where there is a clinical suspicion of cardiomyopathy with or without the following manifestations: - Shortness of breath - Fatigue - Arrythmia (abnormal heart rhythm) - Family history of arrhythmia - Abnormal scans - Ventricular tachycardia - Ventricular fibrillation - Chest Pain - Dizziness - Sudden cardiac death in the family 1 Clinical Utility The clinical utility of this panel is: - The genetic and molecular diagnosis for an accurate clinical diagnosis of a patient with personal or family history of cardiomyopathy, channelopathy or sudden cardiac death. -

Essential Role of Autophagy in Protecting Neonatal Haematopoietic Stem Cells from Oxidative Stress in a P62-Independent Manner
www.nature.com/scientificreports OPEN Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62‑independent manner Naho Nomura1,7,10, Chiaki Ito1,10, Takako Ooshio1,8, Yuko Tadokoro1,2, Susumu Kohno3, Masaya Ueno1,2, Masahiko Kobayashi1,2, Atsuko Kasahara4, Yusuke Takase1,9, Kenta Kurayoshi1, Sha Si1,2, Chiaki Takahashi3, Masaaki Komatsu5, Toru Yanagawa6 & Atsushi Hirao1,2* Autophagy is a cellular degradation system contributing to homeostasis of tissue stem cells including haematopoietic stem cells (HSCs). It plays pleiotropic roles in HSC characteristics throughout life, but its stage‑specifc roles in HSC self‑renewal are unclear. To investigate the efects of Atg5 deletion on stage‑specifc HSC functions, we compared the repopulating capacity of HSCs in Atg5f/f;Vavi-cre mice from postnatal day (P) 0–7 weeks of age. Interestingly, Atg5 defciency led to no remarkable abnormality in the HSC self‑renewal capacity at P0, but signifcant defects at P7, followed by severe defects. Induction of Atg5 deletion at P5 by tamoxifen administration to Atg5f/f;Rosa26-Cre-ERT2 mice resulted in normal haematopoiesis, including the HSC population, until around 1 year, suggesting that Atg5 in the early neonatal period was critical for haematopoiesis in adults. Mitochondrial oxidative stress was increased by Atg5 loss in neonatal HSC/progenitor cells. Although p62 had accumulated in immature bone marrow cells of Atg5f/f;Vavi-cre mice, p62 deletion did not restore defective HSC functions, indicating that Atg5‑dependent haematopoietic regulation in the developmental period was independent of p62. This study proposes a critical role of autophagy in HSC protection against harsh environments in the early neonatal stage, which is essential for healthy long‑term haematopoiesis. -

Whole Exome Sequencing Gene Package Intellectual Disability, Version 9.1, 31-1-2020
Whole Exome Sequencing Gene package Intellectual disability, version 9.1, 31-1-2020 Technical information DNA was enriched using Agilent SureSelect DNA + SureSelect OneSeq 300kb CNV Backbone + Human All Exon V7 capture and paired-end sequenced on the Illumina platform (outsourced). The aim is to obtain 10 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate and non-unique reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA-MEM algorithm (reference: http://bio-bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x A2ML1 {Otitis media, susceptibility to}, 166760 610627 66 100 100 96 AARS1 Charcot-Marie-Tooth disease, axonal, type 2N, 613287 601065 63 100 97 90 Epileptic encephalopathy, early infantile, 29, 616339 AASS Hyperlysinemia, -

Noonan Spectrum Disorders Panel, Sequencing ARUP Test Code 2010772 Noonan Disorders Sequencing Specimen Whole Blood
Patient Report |FINAL Client: Example Client ABC123 Patient: Patient, Example 123 Test Drive Salt Lake City, UT 84108 DOB 12/9/2011 UNITED STATES Gender: Female Patient Identifiers: 01234567890ABCD, 012345 Physician: Doctor, Example Visit Number (FIN): 01234567890ABCD Collection Date: 01/01/2017 12:34 Noonan Spectrum Disorders Panel, Sequencing ARUP test code 2010772 Noonan Disorders Sequencing Specimen Whole Blood Noonan Disorders Sequencing Interp Positive INDICATION FOR TESTING Short stature and facial features suggestive of Noonan syndrome. RESULT One pathogenic variant was detected in the PTPN11 gene. PATHOGENIC VARIANT Gene: PTPN11 (NM_002834.3) Nucleic Acid Change: c.188A>G; Heterozygous Amino Acid Alteration: p.Tyr63Cys Inheritance: Autosomal Dominant INTERPRETATION One pathogenic variant, c.188A>G; p.Tyr63Cys, was detected in the PTPN11 gene by massively parallel sequencing and confirmed by Sanger sequencing. Pathogenic PTPN11 variants are inherited in an autosomal dominant manner, and are associated with Noonan syndrome 1 (MIM: 163950), metachondromatosis (MIM: 156250), and LEOPARD syndrome 1 (MIM: 151100). No additional pathogenic variants were identified in the other targeted genes by massively parallel sequencing. Please refer to the background information included in this report for a list of the genes analyzed and limitations of this test. Evidence for variant classification: The PTPN11 c.188A>G, p.Tyr63Cys variant (rs121918459) has been reported in multiple patients diagnosed with Noonan syndrome (Tartaglia 2001, Jongmans 2011, Martinelli 2012, Hashida 2013, Lepri 2014, Okamoto 2015). This variant is located in a structurally important region of the catalytic N-terminal SH2 domain of PTPN11 (Hof 1998), and several additional variants in neighboring codons have also been identified in Noonan patients (Jongmans 2011, Tartaglia 2002, Tartaglia 2006). -

Mir-126-3P Contributes to Sorafenib Resistance in Hepatocellular Carcinoma Via Downregulating SPRED1
38 Original Article Page 1 of 14 miR-126-3p contributes to sorafenib resistance in hepatocellular carcinoma via downregulating SPRED1 Wenliang Tan1,2#, Zhirong Lin1,2#, Xianqing Chen3, Wenxin Li4, Sicong Zhu1,5, Yingcheng Wei1,2, Liyun Huo1,2, Yajin Chen1,2, Changzhen Shang1,2 1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China; 2Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China; 3Department of Hepatobiliary Surgery, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 4Department of Cardiology, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 5Department of Surgical Intensive Care Unit, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China Contributions: (I) Conception and design: C Shang, Y Chen; (II) Administrative support: C Shang, Y Chen; (III) Provision of study materials or patients: W Tan, Z Lin; (IV) Collection and assembly of data: S Zhu, Y Wei, L Huo; (V) Data analysis and interpretation: X Chen, W Li; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Changzhen Shang; Yajin Chen. Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou 510120, China. Email: [email protected]; [email protected]. Background: Sorafenib can prolong the survival of patients with advanced hepatocellular carcinoma (HCC). However, drug resistance remains the main obstacle to improving its efficiency. This study aimed to explore the likely molecular mechanism of sorafenib resistance. Methods: Differentially expressed microRNAs (miRNAs) related to sorafenib response were analyzed with the Limma package in R software. -

Affinity Purification of NF1 Protein–Protein Interactors Identifies
G C A T T A C G G C A T genes Article Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners Rachel M. Carnes 1, Robert A. Kesterson 1 , Bruce R. Korf 1, James A. Mobley 2 and Deeann Wallis 1,* 1 Department of Genetics, The University of Alabama at Birmingham, Birmingham, AL 35294, USA 2 Department of Anesthesiology and Perioperative Medicine, The University of Alabama at Birmingham, Birmingham, AL 35294, USA * Correspondence: [email protected]; Tel.: +1-205-934-2794; Fax: +1-205-975-4418 Received: 8 July 2019; Accepted: 19 August 2019; Published: 28 August 2019 Abstract: Neurofibromatosis Type 1 (NF1) is caused by pathogenic variants in the NF1 gene encoding neurofibromin. Definition of NF1 protein–protein interactions (PPIs) has been difficult and lacks replication, making it challenging to define binding partners that modulate its function. We created a novel tandem affinity purification (TAP) tag cloned in frame to the 3’ end of the full-length murine Nf1 cDNA (mNf1). We show that this cDNA is functional and expresses neurofibromin, His-Tag, and can correct p-ERK/ERK ratios in NF1 null HEK293 cells. We used this affinity tag to purify binding partners with Strep-Tactin®XT beads and subsequently, identified them via mass spectrometry (MS). We found the tagged mNf1 can affinity purify human neurofibromin and vice versa, indicating that neurofibromin oligomerizes. We identify 21 additional proteins with high confidence of interaction with neurofibromin. After Metacore network analysis of these 21 proteins, eight appear within the same network, primarily keratins regulated by estrogen receptors. -
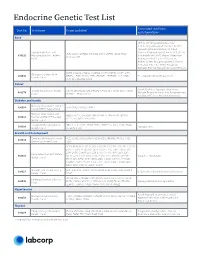
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

Comprehensive Inherited Cancer Precision Panel Overview
Comprehensive Inherited Cancer Precision Panel Overview Hereditary cancer syndromes are encountered in all medical specialties. Although they account for about 5% of all malignancies, it is of special importance to identify these patients because, unlike patients with sporadic cancers, they require special, long-term care as their predisposition can cause them to develop certain tumors at a relatively early age. These cancers can arise in the lungs, kidneys, liver, pancreas, skin, eyes, heart. Most hereditary cancers are associated with a “germline mutation” that will be present in every cell of the human body. Identification of patients at risk of inherited cancer susceptibility is dependent upon the ability to characterize genes and alterations associated with increased cancer risk as well as gathering a detailed personal and family history aiding in the identification of the mode of inheritance as well as other family members at risk of suffering from this susceptibility. Most hereditary cancer syndromes follow an autosomal dominant inheritance, and the penetrance is high. The Igenomix Comprehensive Inherited Cancer Precision Panel provides a comprehensive analysis of the most common hereditary cancer syndromes using next-generation sequencing (NGS) to fully understand the spectrum of relevant cancer predisposition genes. Indications The Igenomix Comprehensive Inherited Caner Precision Panel is indicated as a screening and diagnostic test in those cases where there are: ‐ Multiple relatives on the same side of the family with the same or related forms of cancer ‐ Cancer at an early age ‐ Early presentation of an aggressive cancer type ‐ Multiple primary cancers in an individual 1 Clinical Utility The clinical utility of this panel is: ‐ Early and accurate genetic diagnosis allowing the most appropriate clinical management of a patient with personal or family history suggestive of a hereditary cancer syndrome. -

NGS Panels 2020
NGS Panels 2020 BENEFIT FROM OUR MEDICAL EXPERTISE AND STREAMLINED GENETIC TESTING NGS Panels 2020 Benefit from our Medical Expertise and Streamlined Genetic Testing CENTOGENE is fully committed to bringing the best possible diagnostic solutions to our patients and their families. We always strive to incorporate the latest in-house findings and medical research in our products to improve and ease the diagnostic odyssey of rare disease patients. To reflect the fast-growing knowledge of complex associations of genes with diseases as well as to maximize clinical sensitivity, we have updated and significantly redesigned our Next Generation Sequencing (NGS) gene panels. The gene composition of each panel has been revised to meet the latest gene discoveries as well as to provide the highest clinical validity. Additionally, we have minimized complexity and removed redundancy in the panel portfolio by creating phenotype-directed diagnostic panels, which are the most comprehensive and include all relevant genes necessary for differential diagnosis of syndromes with overlapping phenotype, therefore allowing the diagnosis of diseases that otherwise would be missed. This principle increases the clinical utility, de-risks panel choice, increases cost-effectiveness, and ultimately simplifies the diagnostic process. When choosing one of our NGS panels, feel confident that you will receive high-quality sequencing combined with best data analysis and interpretation, which are documented in comprehensive medical reports. As always, CENTOGENE and our Customer -

Blueprint Genetics Noonan Syndrome Panel
Noonan Syndrome Panel Test code: CA0501 Is a 35 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of a RASopathy including Noonan syndrome with or without lentigines, cardio- facio-cutaneous (CFC) syndrome, Costello syndrome, Noonan-like syndromes or other syndromes causing differential diagnostic challenges such as Legius syndrome, Baraitser-Winter syndromes and neurofibromatosis. About Noonan Syndrome Noonan syndrome is one of the most common syndromes with an estimated prevalence of 1 in 1,000 to 1 in 2,500 live births. It is clinically and genetically heterogeneous condition characterized by cardiovascular abnormalities, distinctive facial features, chest deformity, short stature, and other co-morbidities. Among the Noonan syndrome associated genes, many different genotype-phenotype correlations have been established although no phenotypic features are exclusively associated with one genotype. There are, however, significant differences in the risk of various Noonan syndrome manifestations based on the causative gene. Availability 4 weeks Gene Set Description Genes in the Noonan Syndrome Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACTB* Baraitser-Winter syndrome AD 55 60 ACTG1* Deafness, Baraitser-Winter syndrome AD 27 47 BRAF* LEOPARD syndrome, Noonan syndrome, Cardiofaciocutaneous syndrome AD 134 65 CBL Noonan syndrome-like disorder with or without juvenile myelomonocytic AD 24 43 leukemia CCNK Intellectual disability AD CDC42