Piggybac Mutagenesis and Exome Sequencing Identify Genetic Driver
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Essential Role of Autophagy in Protecting Neonatal Haematopoietic Stem Cells from Oxidative Stress in a P62-Independent Manner
www.nature.com/scientificreports OPEN Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62‑independent manner Naho Nomura1,7,10, Chiaki Ito1,10, Takako Ooshio1,8, Yuko Tadokoro1,2, Susumu Kohno3, Masaya Ueno1,2, Masahiko Kobayashi1,2, Atsuko Kasahara4, Yusuke Takase1,9, Kenta Kurayoshi1, Sha Si1,2, Chiaki Takahashi3, Masaaki Komatsu5, Toru Yanagawa6 & Atsushi Hirao1,2* Autophagy is a cellular degradation system contributing to homeostasis of tissue stem cells including haematopoietic stem cells (HSCs). It plays pleiotropic roles in HSC characteristics throughout life, but its stage‑specifc roles in HSC self‑renewal are unclear. To investigate the efects of Atg5 deletion on stage‑specifc HSC functions, we compared the repopulating capacity of HSCs in Atg5f/f;Vavi-cre mice from postnatal day (P) 0–7 weeks of age. Interestingly, Atg5 defciency led to no remarkable abnormality in the HSC self‑renewal capacity at P0, but signifcant defects at P7, followed by severe defects. Induction of Atg5 deletion at P5 by tamoxifen administration to Atg5f/f;Rosa26-Cre-ERT2 mice resulted in normal haematopoiesis, including the HSC population, until around 1 year, suggesting that Atg5 in the early neonatal period was critical for haematopoiesis in adults. Mitochondrial oxidative stress was increased by Atg5 loss in neonatal HSC/progenitor cells. Although p62 had accumulated in immature bone marrow cells of Atg5f/f;Vavi-cre mice, p62 deletion did not restore defective HSC functions, indicating that Atg5‑dependent haematopoietic regulation in the developmental period was independent of p62. This study proposes a critical role of autophagy in HSC protection against harsh environments in the early neonatal stage, which is essential for healthy long‑term haematopoiesis. -

Mir-126-3P Contributes to Sorafenib Resistance in Hepatocellular Carcinoma Via Downregulating SPRED1
38 Original Article Page 1 of 14 miR-126-3p contributes to sorafenib resistance in hepatocellular carcinoma via downregulating SPRED1 Wenliang Tan1,2#, Zhirong Lin1,2#, Xianqing Chen3, Wenxin Li4, Sicong Zhu1,5, Yingcheng Wei1,2, Liyun Huo1,2, Yajin Chen1,2, Changzhen Shang1,2 1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China; 2Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China; 3Department of Hepatobiliary Surgery, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 4Department of Cardiology, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 5Department of Surgical Intensive Care Unit, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China Contributions: (I) Conception and design: C Shang, Y Chen; (II) Administrative support: C Shang, Y Chen; (III) Provision of study materials or patients: W Tan, Z Lin; (IV) Collection and assembly of data: S Zhu, Y Wei, L Huo; (V) Data analysis and interpretation: X Chen, W Li; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Changzhen Shang; Yajin Chen. Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou 510120, China. Email: [email protected]; [email protected]. Background: Sorafenib can prolong the survival of patients with advanced hepatocellular carcinoma (HCC). However, drug resistance remains the main obstacle to improving its efficiency. This study aimed to explore the likely molecular mechanism of sorafenib resistance. Methods: Differentially expressed microRNAs (miRNAs) related to sorafenib response were analyzed with the Limma package in R software. -

Affinity Purification of NF1 Protein–Protein Interactors Identifies
G C A T T A C G G C A T genes Article Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners Rachel M. Carnes 1, Robert A. Kesterson 1 , Bruce R. Korf 1, James A. Mobley 2 and Deeann Wallis 1,* 1 Department of Genetics, The University of Alabama at Birmingham, Birmingham, AL 35294, USA 2 Department of Anesthesiology and Perioperative Medicine, The University of Alabama at Birmingham, Birmingham, AL 35294, USA * Correspondence: [email protected]; Tel.: +1-205-934-2794; Fax: +1-205-975-4418 Received: 8 July 2019; Accepted: 19 August 2019; Published: 28 August 2019 Abstract: Neurofibromatosis Type 1 (NF1) is caused by pathogenic variants in the NF1 gene encoding neurofibromin. Definition of NF1 protein–protein interactions (PPIs) has been difficult and lacks replication, making it challenging to define binding partners that modulate its function. We created a novel tandem affinity purification (TAP) tag cloned in frame to the 3’ end of the full-length murine Nf1 cDNA (mNf1). We show that this cDNA is functional and expresses neurofibromin, His-Tag, and can correct p-ERK/ERK ratios in NF1 null HEK293 cells. We used this affinity tag to purify binding partners with Strep-Tactin®XT beads and subsequently, identified them via mass spectrometry (MS). We found the tagged mNf1 can affinity purify human neurofibromin and vice versa, indicating that neurofibromin oligomerizes. We identify 21 additional proteins with high confidence of interaction with neurofibromin. After Metacore network analysis of these 21 proteins, eight appear within the same network, primarily keratins regulated by estrogen receptors. -
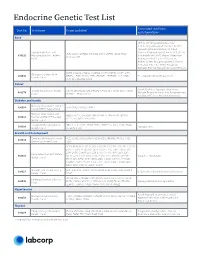
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

Comprehensive Inherited Cancer Precision Panel Overview
Comprehensive Inherited Cancer Precision Panel Overview Hereditary cancer syndromes are encountered in all medical specialties. Although they account for about 5% of all malignancies, it is of special importance to identify these patients because, unlike patients with sporadic cancers, they require special, long-term care as their predisposition can cause them to develop certain tumors at a relatively early age. These cancers can arise in the lungs, kidneys, liver, pancreas, skin, eyes, heart. Most hereditary cancers are associated with a “germline mutation” that will be present in every cell of the human body. Identification of patients at risk of inherited cancer susceptibility is dependent upon the ability to characterize genes and alterations associated with increased cancer risk as well as gathering a detailed personal and family history aiding in the identification of the mode of inheritance as well as other family members at risk of suffering from this susceptibility. Most hereditary cancer syndromes follow an autosomal dominant inheritance, and the penetrance is high. The Igenomix Comprehensive Inherited Cancer Precision Panel provides a comprehensive analysis of the most common hereditary cancer syndromes using next-generation sequencing (NGS) to fully understand the spectrum of relevant cancer predisposition genes. Indications The Igenomix Comprehensive Inherited Caner Precision Panel is indicated as a screening and diagnostic test in those cases where there are: ‐ Multiple relatives on the same side of the family with the same or related forms of cancer ‐ Cancer at an early age ‐ Early presentation of an aggressive cancer type ‐ Multiple primary cancers in an individual 1 Clinical Utility The clinical utility of this panel is: ‐ Early and accurate genetic diagnosis allowing the most appropriate clinical management of a patient with personal or family history suggestive of a hereditary cancer syndrome. -

Feedback Regulation of RTK Signaling in Development ⁎ Cynthia L
Developmental Biology xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Developmental Biology journal homepage: www.elsevier.com/locate/developmentalbiology Review article Feedback regulation of RTK signaling in development ⁎ Cynthia L. Nebena, Megan Loa,b, Natalia Jurab,c, , Ophir D. Kleina,d,⁎⁎ a Department of Orofacial Sciences and Program in Craniofacial Biology, University of California, San Francisco, San Francisco 94143, USA b Cardiovascular Research Institute, University of California, San Francisco, San Francisco, CA, USA c Department of Cellular and Molecular Pharmacology, University of California, San Francisco, San Francisco, CA, USA d Department of Pediatrics and Institute for Human Genetics, University of California, San Francisco, San Francisco 94143, USA ARTICLE INFO ABSTRACT Keywords: Precise regulation of the amplitude and duration of receptor tyrosine kinase (RTK) signaling is critical for the Receptor tyrosine kinases execution of cellular programs and behaviors. Understanding these control mechanisms has important Signaling pathways implications for the field of developmental biology, and in recent years, the question of how augmentation or Feedback regulation attenuation of RTK signaling via feedback loops modulates development has become of increasing interest. RTK Developmental control feedback regulation is also important for human disease research; for example, germline mutations in genes RASopathies that encode RTK signaling pathway components cause numerous human congenital syndromes, and somatic Sprouty alterations contribute to the pathogenesis of diseases such as cancers. In this review, we survey regulators of RTK signaling that tune receptor activity and intracellular transduction cascades, with a focus on the roles of these genes in the developing embryo. We detail the diverse inhibitory mechanisms utilized by negative feedback regulators that, when lost or perturbed, lead to aberrant increases in RTK signaling. -

Live-Cell Imaging Rnai Screen Identifies PP2A–B55α and Importin-Β1 As Key Mitotic Exit Regulators in Human Cells
LETTERS Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells Michael H. A. Schmitz1,2,3, Michael Held1,2, Veerle Janssens4, James R. A. Hutchins5, Otto Hudecz6, Elitsa Ivanova4, Jozef Goris4, Laura Trinkle-Mulcahy7, Angus I. Lamond8, Ina Poser9, Anthony A. Hyman9, Karl Mechtler5,6, Jan-Michael Peters5 and Daniel W. Gerlich1,2,10 When vertebrate cells exit mitosis various cellular structures can contribute to Cdk1 substrate dephosphorylation during vertebrate are re-organized to build functional interphase cells1. This mitotic exit, whereas Ca2+-triggered mitotic exit in cytostatic-factor- depends on Cdk1 (cyclin dependent kinase 1) inactivation arrested egg extracts depends on calcineurin12,13. Early genetic studies in and subsequent dephosphorylation of its substrates2–4. Drosophila melanogaster 14,15 and Aspergillus nidulans16 reported defects Members of the protein phosphatase 1 and 2A (PP1 and in late mitosis of PP1 and PP2A mutants. However, the assays used in PP2A) families can dephosphorylate Cdk1 substrates in these studies were not specific for mitotic exit because they scored pro- biochemical extracts during mitotic exit5,6, but how this relates metaphase arrest or anaphase chromosome bridges, which can result to postmitotic reassembly of interphase structures in intact from defects in early mitosis. cells is not known. Here, we use a live-cell imaging assay and Intracellular targeting of Ser/Thr phosphatase complexes to specific RNAi knockdown to screen a genome-wide library of protein substrates is mediated by a diverse range of regulatory and targeting phosphatases for mitotic exit functions in human cells. We subunits that associate with a small group of catalytic subunits3,4,17. -

The Neurofibromin Recruitment Factor Spred1 Binds to the GAP Related Domain Without Affecting Ras Inactivation
The neurofibromin recruitment factor Spred1 binds to the GAP related domain without affecting Ras inactivation Theresia Dunzendorfer-Matta,1, Ellen L. Mercadob,1, Karl Malyc, Frank McCormickb,2, and Klaus Scheffzeka,2 aDivision of Biological Chemistry, Biocenter, Medical University of Innsbruck, 6020 Innsbruck, Austria; bHelen Diller Family Comprehensive Cancer Center, University of California, San Francisco, CA 94158; and cDivision of Medical Biochemistry, Biocenter, Medical University of Innsbruck, 6020 Innsbruck, Austria Contributed by Frank McCormick, May 10, 2016 (sent for review November 3, 2015; reviewed by Jonathan Licht and Nancy Ratner) Neurofibromatosis type 1 (NF1) and Legius syndrome are related glycerophospholipid binding Sec14-like domain associated with a diseases with partially overlapping symptoms caused by alterations previously undetected C-terminal pleckstrin homology (PH)-like of the tumor suppressor genes NF1 (encoding the protein neurofibro- domain (13–15). Although a number of interaction partners of min) and SPRED1 (encoding sprouty-related, EVH1 domain-containing neurofibromin have been reported, the significance of the un- protein 1, Spred1), respectively. Both proteins are negative regulators derlying protein–protein interaction is not clearly established in of Ras/MAPK signaling with neurofibromin functioning as a Ras- many cases, as reviewed in refs. 2 and 16. specific GTPase activating protein (GAP) and Spred1 acting on hitherto The recently described Legius syndrome (LS) shares the undefined components of the pathway. Importantly, neurofibromin milder features with NF1, including café au lait macules, axillary has been identified as a key protein in the development of cancer, as freckling, and sometimes macrocephaly, but does not appear to it is genetically altered in a large number of sporadic human malig- include the more severe clinical manifestations of NF1 such as nancies unrelated to NF1. -

Comprehensive Testing for NF1/SPRED1 and Other Rasopathies Genes at UAB
Medical Genomics Laboratory Department of Genetics Noonan syndrome/NSML Costello syndrome Cardio-facio-cutaneous syndrome Neurofibromatosis type 1 Legius syndrome Comprehensive Testing for NF1/SPRED1 and Other RASopathies Genes at UAB 720 20th Street S | KAUL Building - Suite 330 | Birmingham, Alabama 35294 Phone: 205-934-5562 | Fax: 205-996-2929 | email: [email protected] Testing for NF1/SPRED1 and other RASopathies genes at UAB a comprehensive menu allowing a tailored approach for patients with constitutional or mosaic presentations NF1-only NF1, SPRED1, PTPN11, NF1-SPRED1 DNA-based testing by NGS BRAF, CBL, HRAS, KRAS, (effective April 18th, 2016) Expanded NF1-RASopathy (16 genes) NRAS, MAP2K1, MAP2K2, non-NF1 RASopathy (15 genes) RAF1, RIT1, RASA2, SHOC2, RNA-based testing by Sanger Comprehensive NF1/SPRED1 testing SOS1 and SOS2 (16 genes) on blood and affected tissues BACKGROUND The RASopathies are a genetically heterogeneous group of disorders caused by mutations in the genes involved in the Ras-MAPK pathway. As a group, the RASopathies are one of the largest groups of malforma- tion syndromes known, affecting ~1:1,000 and include Neurofibromatosis type 1, Legius syndrome, Noonan syndrome, cardio-facio-cutaneous (CFC) syndrome, Noonan Syndrome with Multiple Lentigines (NSML/- LEOPARD) and Costello syndrome. Mutations in NF1 and SPRED1 are typically loss-of-function mutations and include the full spectrum of nonsense, missense, splice, frameshift, insertion-deletion, and copy number changes. Mutations in the other RASopathy genes are typically missense mutations or/and in-frame deletion/insertion of an amino acid. The Ras/MAPK pathway can have a profound deleterious effect on development as it plays a key role in differentiation, growth, senescence, and dysregulation. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

The MAPK Pathway As an Apoptosis Enhancer in Melanoma
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 5, No. 13 The MAPK pathway as an apoptosis enhancer in melanoma Johannes M. Haydn1, Anita Hufnagel1, Johannes Grimm1, Katja Maurus1, Manfred Schartl1,2 and Svenja Meierjohann1,2 1 Department of Physiological Chemistry, Biocenter, University of Wurzburg, Wurzburg, Germany 2 Comprehensive Cancer Center Mainfranken, University Hospital Wurzburg, Germany Correspondence to: Svenja Meierjohann, email: [email protected] Keywords: Crosstalk, chemotherapy resistance, RAS, PI3K, melanoma Received: March 18, 2014 Accepted: June 6, 2014 Published: June 8, 2014 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Inhibition of RAF/MEK/ERK signaling is beneficial for many patients with BRAFV600E–mutated melanoma. However, primary and secondary resistances restrict long-lasting therapy success. Combination therapies are therefore urgently needed. Here, we evaluate the cellular effect of combining a MEK inhibitor with a genotoxic apoptosis inducer. Strikingly, we observed that an activated MAPK pathway promotes in several melanoma cell lines the pro-apoptotic response to genotoxic stress, and MEK inhibition reduces intrinsic apoptosis. This goes along with MEK inhibitor induced increased RAS and P-AKT levels. The protective effect of the MEK inhibitor depends on PI3K signaling, which prevents the induction of pro-apoptotic PUMA that mediates apoptosis after DNA damage. We could show that the MEK inhibitor dependent feedback loop is enabled by several factors, including EGF receptor and members of the SPRED family. The simultaneous knockdown of SPRED1 and SPRED2 mimicked the effects of MEK inhibitor such as PUMA repression and protection from apoptosis. -

Frizzled7: a Promising Achilles' Heel for Targeting the Wnt Receptor
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Online Research @ Cardiff cancers Review Frizzled7: A Promising Achilles’ Heel for Targeting the Wnt Receptor Complex to Treat Cancer Toby Phesse 1,2,*,†,‡, Dustin Flanagan 1,† and Elizabeth Vincan 1,3,* 1 Molecular Oncology Laboratory, Victorian Infectious Diseases Reference Laboratory and the Doherty Institute, University of Melbourne, Melbourne, VIC 3000, Australia; dustin.fl[email protected] 2 Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC 3052, Australia 3 School of Biomedical Sciences, Curtin University, Perth, WA 6102, Australia * Correspondence: [email protected] (T.P.); [email protected] (E.V.); Tel.: +44-29-2087-4089 (T.P.); +61-(0)-9342-9348 (E.V.) † These authors contributed equally to this work. ‡ Current Address: European Cancer Stem Cell Research Institute, Cardiff University, Cardiff, CF24 4HQ, UK. Academic Editors: Renée van Amerongen and Walter Birchmeier Received: 31 March 2016; Accepted: 9 May 2016; Published: 17 May 2016 Abstract: Frizzled7 is arguably the most studied member of the Frizzled family, which are the cognate Wnt receptors. Frizzled7 is highly conserved through evolution, from Hydra through to humans, and is expressed in diverse organisms, tissues and human disease contexts. Frizzled receptors can homo- or hetero-polymerise and associate with several co-receptors to transmit Wnt signalling. Notably, Frizzled7 can transmit signalling via multiple Wnt transduction pathways and bind to several different Wnt ligands, Frizzled receptors and co-receptors. These promiscuous binding and functional properties are thought to underlie the pivotal role Frizzled7 plays in embryonic developmental and stem cell function.