Affinity Purification of NF1 Protein–Protein Interactors Identifies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Piggybac Mutagenesis and Exome Sequencing Identify Genetic Driver
Noorani et al. Genome Biology (2020) 21:181 https://doi.org/10.1186/s13059-020-02092-2 RESEARCH Open Access PiggyBac mutagenesis and exome sequencing identify genetic driver landscapes and potential therapeutic targets of EGFR-mutant gliomas Imran Noorani1,2* , Jorge de la Rosa1†, Yoon Ha Choi1,3†, Alexander Strong1, Hannes Ponstingl1, M. S. Vijayabaskar1, Jusung Lee3, Eunmin Lee3, Angela Richard-Londt4, Mathias Friedrich1,5, Federica Furlanetto5, Rocio Fuente1, Ruby Banerjee1, Fengtang Yang1, Frances Law1, Colin Watts2,6, Roland Rad5, George Vassiliou1, Jong Kyoung Kim3, Thomas Santarius2, Sebastian Brandner4 and Allan Bradley1* * Correspondence: [email protected]; [email protected] Abstract †Jorge de la Rosa and Yoonha Choi contributed equally to this work. Background: Glioma is the most common intrinsic brain tumor and also occurs in 1The Wellcome Trust Sanger the spinal cord. Activating EGFR mutations are common in IDH1 wild-type gliomas. Institute, Wellcome Trust Genome However, the cooperative partners of EGFR driving gliomagenesis remain poorly Campus, Hinxton, Cambridgeshire CB10 1SA, UK understood. Full list of author information is Results: We explore EGFR-mutant glioma evolution in conditional mutant mice by available at the end of the article whole-exome sequencing, transposon mutagenesis forward genetic screening, and transcriptomics. We show mutant EGFR is sufficient to initiate gliomagenesis in vivo, both in the brain and spinal cord. We identify significantly recurrent somatic alterations in these gliomas including mutant EGFR amplifications and Sub1, Trp53, and Tead2 loss-of-function mutations. Comprehensive functional characterization of 96 gliomas by genome-wide piggyBac insertional mutagenesis in vivo identifies 281 known and novel EGFR-cooperating driver genes, including Cdkn2a, Nf1, Spred1, and Nav3. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Essential Role of Autophagy in Protecting Neonatal Haematopoietic Stem Cells from Oxidative Stress in a P62-Independent Manner
www.nature.com/scientificreports OPEN Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62‑independent manner Naho Nomura1,7,10, Chiaki Ito1,10, Takako Ooshio1,8, Yuko Tadokoro1,2, Susumu Kohno3, Masaya Ueno1,2, Masahiko Kobayashi1,2, Atsuko Kasahara4, Yusuke Takase1,9, Kenta Kurayoshi1, Sha Si1,2, Chiaki Takahashi3, Masaaki Komatsu5, Toru Yanagawa6 & Atsushi Hirao1,2* Autophagy is a cellular degradation system contributing to homeostasis of tissue stem cells including haematopoietic stem cells (HSCs). It plays pleiotropic roles in HSC characteristics throughout life, but its stage‑specifc roles in HSC self‑renewal are unclear. To investigate the efects of Atg5 deletion on stage‑specifc HSC functions, we compared the repopulating capacity of HSCs in Atg5f/f;Vavi-cre mice from postnatal day (P) 0–7 weeks of age. Interestingly, Atg5 defciency led to no remarkable abnormality in the HSC self‑renewal capacity at P0, but signifcant defects at P7, followed by severe defects. Induction of Atg5 deletion at P5 by tamoxifen administration to Atg5f/f;Rosa26-Cre-ERT2 mice resulted in normal haematopoiesis, including the HSC population, until around 1 year, suggesting that Atg5 in the early neonatal period was critical for haematopoiesis in adults. Mitochondrial oxidative stress was increased by Atg5 loss in neonatal HSC/progenitor cells. Although p62 had accumulated in immature bone marrow cells of Atg5f/f;Vavi-cre mice, p62 deletion did not restore defective HSC functions, indicating that Atg5‑dependent haematopoietic regulation in the developmental period was independent of p62. This study proposes a critical role of autophagy in HSC protection against harsh environments in the early neonatal stage, which is essential for healthy long‑term haematopoiesis. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

Views, What Controversies Remain and Why We Still 39
BMC Cancer BioMed Central Research article Open Access Heterogeneous in vitro effects of doxorubicin on gene expression in primary human liposarcoma cultures Adrien Daigeler*1, Ludger Klein-Hitpass2, Michael Ansgar Chromik3, Oliver Müller4, Jörg Hauser1, Heinz-Herbert Homann1, Hans-Ulrich Steinau1 and Marcus Lehnhardt1 Address: 1Department of Plastic Surgery, Burn Center, Hand surgery, Sarcoma Reference Center, BG-University Hospital Bergmannsheil, Ruhr University Bochum, Bürkle-de-la-Camp-Platz 1, 44789 Bochum, Germany, 2Institute of Cell Biology (Tumor Research), IFZ, University of Essen, Virchowstr. 173, 45122 Essen, Germany, 3Department of Surgery, St. Josef Hospital, Ruhr-University, Bochum, Germany and 4Tumor Genetics Group, Max-Planck-Institut für molekulare Physiologie, Otto Hahnstr. 11, 44227 Dortmund, Germany Email: Daigeler Adrien* - [email protected]; Klein-Hitpass Ludger - [email protected]; Chromik Ansgar Michael - [email protected]; Müller Oliver - [email protected]; Hauser Jörg - [email protected]; Homann Heinz-Herbert - [email protected]; Steinau Hans-Ulrich - hans- [email protected]; Lehnhardt Marcus - [email protected] * Corresponding author Published: 29 October 2008 Received: 17 December 2007 Accepted: 29 October 2008 BMC Cancer 2008, 8:313 doi:10.1186/1471-2407-8-313 This article is available from: http://www.biomedcentral.com/1471-2407/8/313 © 2008 Adrien et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Doxorubicin is considered one of the most potent established chemotherapeutics in the treatment of liposarcoma; however, the response rates usually below 30%, are still disappointing. -

Mir-126-3P Contributes to Sorafenib Resistance in Hepatocellular Carcinoma Via Downregulating SPRED1
38 Original Article Page 1 of 14 miR-126-3p contributes to sorafenib resistance in hepatocellular carcinoma via downregulating SPRED1 Wenliang Tan1,2#, Zhirong Lin1,2#, Xianqing Chen3, Wenxin Li4, Sicong Zhu1,5, Yingcheng Wei1,2, Liyun Huo1,2, Yajin Chen1,2, Changzhen Shang1,2 1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China; 2Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China; 3Department of Hepatobiliary Surgery, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 4Department of Cardiology, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 5Department of Surgical Intensive Care Unit, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China Contributions: (I) Conception and design: C Shang, Y Chen; (II) Administrative support: C Shang, Y Chen; (III) Provision of study materials or patients: W Tan, Z Lin; (IV) Collection and assembly of data: S Zhu, Y Wei, L Huo; (V) Data analysis and interpretation: X Chen, W Li; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Changzhen Shang; Yajin Chen. Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou 510120, China. Email: [email protected]; [email protected]. Background: Sorafenib can prolong the survival of patients with advanced hepatocellular carcinoma (HCC). However, drug resistance remains the main obstacle to improving its efficiency. This study aimed to explore the likely molecular mechanism of sorafenib resistance. Methods: Differentially expressed microRNAs (miRNAs) related to sorafenib response were analyzed with the Limma package in R software. -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/). -

Evidence for the Role of Ywha in Mouse Oocyte Maturation
EVIDENCE FOR THE ROLE OF YWHA IN MOUSE OOCYTE MATURATION A thesis submitted To Kent State University in partial Fulfillment of the requirements for the Degree of Master of Science By Ariana Claire Detwiler August, 2015 © Copyright All rights reserved Except for previously published materials Thesis written by Ariana Claire Detwiler B.S., Pennsylvania State University, 2012 M.S., Kent State University, 2015 Approved by ___________________________________________________________ Douglas W. Kline, Professor, Ph.D., Department of Biological Sciences, Masters Advisor ___________________________________________________________ Laura G. Leff, Professor, PhD., Chair, Department of Biological Sciences ___________________________________________________________ James L. Blank, Professor, Dean, College of Arts and Sciences i TABLE OF CONTENTS List of Figures ……………………………………………………………………………………v List of Tables ……………………………………………………………………………………vii Acknowledgements …………………………………………………………………………….viii Abstract ……………………………………………………………………………………….....1 Chapter I Introduction…………………………………………………………………………………..2 1.1 Introduction …………………………………………………………………………..2 1.2 Ovarian Function ……………………………………………………………………..2 1.3 Oogenesis and Folliculogenesis ………………………………………………………3 1.4 Oocyte Maturation ……………………………………………………………………5 1.5 Maternal to Embryonic Messenger RNA Transition …………………………………8 1.6 Meiotic Spindle Formation …………………………………………………………...9 1.7 YWHA Isoforms and Oocyte Maturation …………………………………………...10 Aim…………………………………………………………………………………………..15 Chapter II Methods……………………………………………………………………………………...16 -
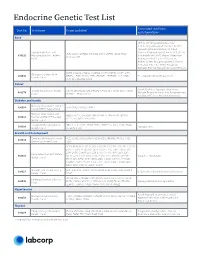
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

Gene Expression Profiles Reveal Alternative Targets of Therapeutic Intervention for the Treatment of Drug-Resistant Non-Small Cell Lung Cancers
University of Kentucky UKnowledge Theses and Dissertations--Pharmacy College of Pharmacy 2017 GENE EXPRESSION PROFILES REVEAL ALTERNATIVE TARGETS OF THERAPEUTIC INTERVENTION FOR THE TREATMENT OF DRUG-RESISTANT NON-SMALL CELL LUNG CANCERS Madeline J. Krentz Gober University of Kentucky, [email protected] Author ORCID Identifier: https://orcid.org/0000-0001-7761-6741 Digital Object Identifier: https://doi.org/10.13023/ETD.2017.309 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Krentz Gober, Madeline J., "GENE EXPRESSION PROFILES REVEAL ALTERNATIVE TARGETS OF THERAPEUTIC INTERVENTION FOR THE TREATMENT OF DRUG-RESISTANT NON-SMALL CELL LUNG CANCERS" (2017). Theses and Dissertations--Pharmacy. 78. https://uknowledge.uky.edu/pharmacy_etds/78 This Doctoral Dissertation is brought to you for free and open access by the College of Pharmacy at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Pharmacy by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Comprehensive Inherited Cancer Precision Panel Overview
Comprehensive Inherited Cancer Precision Panel Overview Hereditary cancer syndromes are encountered in all medical specialties. Although they account for about 5% of all malignancies, it is of special importance to identify these patients because, unlike patients with sporadic cancers, they require special, long-term care as their predisposition can cause them to develop certain tumors at a relatively early age. These cancers can arise in the lungs, kidneys, liver, pancreas, skin, eyes, heart. Most hereditary cancers are associated with a “germline mutation” that will be present in every cell of the human body. Identification of patients at risk of inherited cancer susceptibility is dependent upon the ability to characterize genes and alterations associated with increased cancer risk as well as gathering a detailed personal and family history aiding in the identification of the mode of inheritance as well as other family members at risk of suffering from this susceptibility. Most hereditary cancer syndromes follow an autosomal dominant inheritance, and the penetrance is high. The Igenomix Comprehensive Inherited Cancer Precision Panel provides a comprehensive analysis of the most common hereditary cancer syndromes using next-generation sequencing (NGS) to fully understand the spectrum of relevant cancer predisposition genes. Indications The Igenomix Comprehensive Inherited Caner Precision Panel is indicated as a screening and diagnostic test in those cases where there are: ‐ Multiple relatives on the same side of the family with the same or related forms of cancer ‐ Cancer at an early age ‐ Early presentation of an aggressive cancer type ‐ Multiple primary cancers in an individual 1 Clinical Utility The clinical utility of this panel is: ‐ Early and accurate genetic diagnosis allowing the most appropriate clinical management of a patient with personal or family history suggestive of a hereditary cancer syndrome. -

Exosomes Involved in Cholesterol Metabolism Process of Patients with Spinal Cord Injury in the Acute Phase
ORIGINAL RESEARCH published: 09 July 2021 doi: 10.3389/fninf.2021.662967 Bioinformatic Analysis of the Proteome in Exosomes Derived From Plasma: Exosomes Involved in Cholesterol Metabolism Process of Patients With Spinal Cord Injury in the Acute Phase Chunshuai Wu 1†, Jinjuan Yu 2†, Guanhua Xu 1, Hong Gao 1, Yue Sun 1, Jiayi Huang 1, Li Sun 1, Xu Zhang 1 and Zhiming Cui 1* 1 Department of Spine Surgery, Nantong First People’s Hospital, The Affiliated Hospital 2 of Nantong University, Nantong, China, 2 Department of Administrative Office, The Third People’s Hospital of Nantong, Nantong, China Edited by: Sandeep Kumar Dhanda, St. Jude Children’s Research Hospital, Spinal cord injury (SCI) is a common but severe disease caused by traffic accidents. United States Coronary atherosclerotic heart disease (CHD) caused by dyslipidemia is known as the Reviewed by: leading cause of death in patients with SCI. However, the quantitative analysis showed Prashanth N. Suravajhala, Birla Institute of Scientific that the cholesterol and lipoprotein concentrations in peripheral blood (PB) did not Research, India change significantly within 48 h after SCI. Due to the presence of the Blood spinal Chandrabose Selvaraj, Alagappa University, India cord barrier (BSCB), there are only few studies concerning the plasma cholesterol Aditya Ambati, metabolism in the acute phase of SCI. Exosomes have a smaller particle size, which Stanford University, United States enables them relatively less limitation of BSCB. This study uses exosomes derived from *Correspondence: the plasma of 43 patients in the acute phase of SCI and 71 patients in the control Zhiming Cui [email protected] group as samples.