Whole Exome Sequencing Gene Package Intellectual Disability, Version 9.1, 31-1-2020
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Moderate the MAOA-L Allele Expression with CRISPR/Cas9 System
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 23 April 2018 doi:10.20944/preprints201804.0275.v1 1 Review 2 Moderate the MAOA-L Allele Expression with CRISPR/Cas9 System 3 Martin L. Nelwan 4 Department of Animal Science – Other 5 Nelwan Institution for Human Resource Development 6 Jl. A. Yani No. 24 7 Palu, Sulawesi Tengah, Indonesia 8 Email: [email protected] 9 Abstract: Antisocial behavior is a behavior disorder inherited according to the inheritance of X-linked 10 chromosome. Mutations in the MAOA gene can cause different behaviors in humans. These can comprise 11 violent behavior or antisocial behavior. Low MAOA (MAOA-L) allele activity can cause antisocial 12 behavior in both healthy and unhealthy people. Antisocial from healthy males can originate from 13 maltreatment during childhood. There are no drugs for the treatment of antisocial behavior permanently 14 at this time. MAOA inhibitor can reverse antisocial behavior in animal models. To cure antisocial 15 behavior in the future, the CRISPR/Cas9 system in combination with iPSCs or ssODN methods for 16 instance can be used. This system has succeeded to correct erroneous segments in the F8 gene and F9 17 gene. Both genes occupy the X chromosome. The MAOA gene also occupies the X chromosome. It seems 18 that CRISPR/Cas9 system may be a beneficial tool to edit erroneous segments in the MAOA gene to treat 19 antisocial behavior. 20 Keywords: advanced therapy, aggressive, antisocial, behavior, MAOA. 21 22 1. Introduction 23 Antisocial behavior is a hereditary disorder inherited through an X-linked recessive inheritance 24 pattern. -

Blueprint Genetics Hereditary Leukemia Panel
Hereditary Leukemia Panel Test code: ON0101 Is a 41 gene panel that includes assessment of non-coding variants. Is ideal for patients with a personal history of a syndrome that confers an increased risk of leukemia or patients with a family history of a syndrome that confers an increased risk of leukemia. About Hereditary Leukemia An inherited predisposition to hematological malignancies, namely acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and bone marrow myelodysplastic syndrome (MDS) may be associated with syndromic features or occur as the principal clinical feature. MDSs and AMLs can occur in the context of syndromic bone marrow failure (eg. dyskeratosis congenita, Fanconi anemia). Other hereditary syndromes with an increased risk of leukemia include Li-Fraumeni syndrome (TP53), ataxia telangiectasia (ATM), Bloom syndrome (BLM), neurofibromatosis type 1 (NF1) and less frequently Noonan syndrome (PTPN11, CBL). Some reports have also shown an association of biallelic germline mutations in constitutional mismatch repair-deficiency syndrome genes, MLH1, MSH2, MSH6, and PMS2 with the development of ALL. Isolated hematological malignancies are associated with germline mutations in RUNX1 (familial platelet syndrome with predisposition to acute myelogenous leukemia), CEBPA (familial AML), GATA2 (GATA2-associated syndromes) and DDX41(DDX41 -related myeloid neoplasms). There is a rapidly expanding list of germline mutations associated with increased risks for myeloid malignancies and inherited predisposition to hematologic malignancies may be more common than has been thought. Many different genetic defects associated with the development of leukemia have been described but the common underlying mechanism is a dysfunctional DNA damage response. Recognition of an inherited cause provides a specific molecular diagnosis and helps to guide treatment, understand unique disease features, prognosis and other organ systems that may be involved, and identify others in the family who may be at risk. -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Exostoses, Enchondromatosis and Metachondromatosis; Diagnosis and Management
Acta Orthop. Belg., 2016, 82, 102-105 ORIGINAL STUDY Exostoses, enchondromatosis and metachondromatosis; diagnosis and management John MCFARLANE, Tim KNIGHT, Anubha SINHA, Trevor COLE, Nigel KIELY, Rob FREEMAN From the Department of Orthopaedics, Robert Jones Agnes Hunt Hospital, Oswestry, UK We describe a 5 years old girl who presented to the region of long bones and are composed of a carti- multidisciplinary skeletal dysplasia clinic following lage lump outside the bone which may be peduncu- excision of two bony lumps from her fingers. Based on lated or sessile, the knee is the most common clinical examination, radiolographs and histological site (1,10). An isolated exostosis is a common inci- results an initial diagnosis of hereditary multiple dental finding rarely requiring treatment. Disorders exostosis (HME) was made. Four years later she developed further lumps which had the radiological associated with exostoses include HME, Langer- appearance of enchondromas. The appearance of Giedion syndrome, Gardner syndrome and meta- both exostoses and enchondromas suggested a possi- chondromatosis. ble diagnosis of metachondromatosis. Genetic testing Enchondroma are the second most common be- revealed a splice site mutation at the end of exon 11 on nign bone tumour characterised by the formation of the PTPN11 gene, confirming the diagnosis of meta- hyaline cartilage in the medulla of a bone. It occurs chondromatosis. While both single or multiple exosto- most frequently in the hand (60%) and then the feet. ses and enchondromas occur relatively commonly on The typical radiological features are of a well- their own, the appearance of multiple exostoses and defined lucent defect with endosteal scalloping and enchondromas together is rare and should raise the differential diagnosis of metachondromatosis. -

Preconception Carrier Screening by Genome Sequencing: Results from the Clinical Laboratory
The American Journal of Human Genetics, Volume 102 Supplemental Data Preconception Carrier Screening by Genome Sequencing: Results from the Clinical Laboratory Sumit Punj, Yassmine Akkari, Jennifer Huang, Fei Yang, Allison Creason, Christine Pak, Amiee Potter, Michael O. Dorschner, Deborah A. Nickerson, Peggy D. Robertson, Gail P. Jarvik, Laura M. Amendola, Jennifer Schleit, Dana Kostiner Simpson, Alan F. Rope, Jacob Reiss, Tia Kauffman, Marian J. Gilmore, Patricia Himes, Benjamin Wilfond, Katrina A.B. Goddard, and C. Sue Richards Supplemental Note: Clinical Report Carrier Results: Four Known Pathogenic Variants Detected. Gene Inheritance Disease Prevalence Variant Classification Pendred Syndrome/ Non- syndromic Autosomal Hearing Loss A c.1246A>C, SLC26A4 1/500 Pathogenic Recessive DFNB4 with (p.Thr416Pro) enlarged vestibular aqueduct Autosomal Spastic ++ c.1045G>A, SPG7 2-6/100,000 Pathogenic Recessive Paraplegia 7 (p.Gly349Ser) 3.7 Autosomal Alpha +++ -α HBA2 1-5/10,000 Pathogenic Recessive Thalassemia (α+- thalassemia) Autosomal Hereditary 1/200 – c.845G>A HFE Pathogenic Recessive Hemochromatosis 1/1000+ (p.Cys282Tyr) +: GeneReviews; ++: Genetics Home Reference; +++: orphan.net – varies with population; A- Generalized prevalence of all deafness and hearing loss Interpretation: A sample from this individual was referred to our laboratory for analysis of Next-Generation Genome Sequencing (NGS) and Sanger confirmation of variants identified in carrier screening for: (1) conditions with significantly shortened lifespan; (2) serious conditions; (3) mild conditions; (4) conditions with unpredictable outcomes: and (5) conditions that begin as adults. One known heterozygous missense variant, c.1246A>C (p.Thr416Pro) (NM_000441.1), was detected in exon 10 of the SLC26A4 gene of this individual by NGS. -

Megalencephaly and Macrocephaly
277 Megalencephaly and Macrocephaly KellenD.Winden,MD,PhD1 Christopher J. Yuskaitis, MD, PhD1 Annapurna Poduri, MD, MPH2 1 Department of Neurology, Boston Children’s Hospital, Boston, Address for correspondence Annapurna Poduri, Epilepsy Genetics Massachusetts Program, Division of Epilepsy and Clinical Electrophysiology, 2 Epilepsy Genetics Program, Division of Epilepsy and Clinical Department of Neurology, Fegan 9, Boston Children’s Hospital, 300 Electrophysiology, Department of Neurology, Boston Children’s Longwood Avenue, Boston, MA 02115 Hospital, Boston, Massachusetts (e-mail: [email protected]). Semin Neurol 2015;35:277–287. Abstract Megalencephaly is a developmental disorder characterized by brain overgrowth secondary to increased size and/or numbers of neurons and glia. These disorders can be divided into metabolic and developmental categories based on their molecular etiologies. Metabolic megalencephalies are mostly caused by genetic defects in cellular metabolism, whereas developmental megalencephalies have recently been shown to be caused by alterations in signaling pathways that regulate neuronal replication, growth, and migration. These disorders often lead to epilepsy, developmental disabilities, and Keywords behavioral problems; specific disorders have associations with overgrowth or abnor- ► megalencephaly malities in other tissues. The molecular underpinnings of many of these disorders are ► hemimegalencephaly now understood, providing insight into how dysregulation of critical pathways leads to ► -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Cardiomyopathy Precision Panel Overview Indications
Cardiomyopathy Precision Panel Overview Cardiomyopathies are a group of conditions with a strong genetic background that structurally hinder the heart to pump out blood to the rest of the body due to weakness in the heart muscles. These diseases affect individuals of all ages and can lead to heart failure and sudden cardiac death. If there is a family history of cardiomyopathy it is strongly recommended to undergo genetic testing to be aware of the family risk, personal risk, and treatment options. Most types of cardiomyopathies are inherited in a dominant manner, which means that one altered copy of the gene is enough for the disease to present in an individual. The symptoms of cardiomyopathy are variable, and these diseases can present in different ways. There are 5 types of cardiomyopathies, the most common being hypertrophic cardiomyopathy: 1. Hypertrophic cardiomyopathy (HCM) 2. Dilated cardiomyopathy (DCM) 3. Restrictive cardiomyopathy (RCM) 4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) 5. Isolated Left Ventricular Non-Compaction Cardiomyopathy (LVNC). The Igenomix Cardiomyopathy Precision Panel serves as a diagnostic and tool ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes. Indications The Igenomix Cardiomyopathy Precision Panel is indicated in those cases where there is a clinical suspicion of cardiomyopathy with or without the following manifestations: - Shortness of breath - Fatigue - Arrythmia (abnormal heart rhythm) - Family history of arrhythmia - Abnormal scans - Ventricular tachycardia - Ventricular fibrillation - Chest Pain - Dizziness - Sudden cardiac death in the family 1 Clinical Utility The clinical utility of this panel is: - The genetic and molecular diagnosis for an accurate clinical diagnosis of a patient with personal or family history of cardiomyopathy, channelopathy or sudden cardiac death. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -
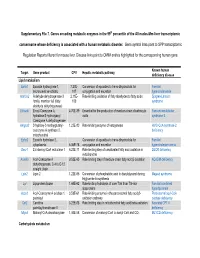
Supplementary File 7. Genes Encoding Metabolic Enzymes in The
Supplementary File 7. Genes encoding metabolic enzymes in the 99th percentile of the All nodes-Mm-liver transcriptomic consensome whose deficiency is associated with a human metabolic disorder. Gene symbol links point to SPP transcriptomic Regulation Reports filtered for mouse liver. Disease links point to OMIM entries highlighted for the corresponding human gene. Known human Target Gene product CPV Hepatic metabolic pathway deficiency disease Lipid metabolism Ephx1 Epoxide hydroxylase 1, 7.27E- Conversion of epoxides to trans-dihydrodiols for Familial microsomal xenobiotic 107 conjugation and excretion hypercholanemia Aldh3a2 Aldehyde dehydrogenase 3 2.11E- Rate-limiting oxidation of fatty aldehydes to fatty acids Sjogren-Larsson family, member A2 (fatty 103 syndrome aldehyde dehydrogenase) Ehhadh Enoyl-Coenzyme A, 4.70E-89 Essential for the production of medium-chain dicarboxylic Fanconi renotubular hydratase/3-hydroxyacyl acids syndrome 3 Coenzyme A dehydrogenase Hmgcs2 3-hydroxy-3-methylglutaryl- 1.23E-80 Rate-limiting enzyme of ketogenesis HMG-CoA synthase-2 coenzyme A synthase 2, deficiency mitochondrial Ephx2 Epoxide hydrolase 2, Conversion of epoxides to trans-dihydrodiols for Familial cytoplasmic 6.06E-78 conjugation and excretion hypercholesterolemia Decr1 2,4-dienoyl-CoA reductase 1 6.23E-71 Rate-limiting step of unsaturated fatty acid oxidation in DECR deficiency mitochondria Acadm Acyl-Coenzyme A 9.52E-65 Rate limiting step of medium-chain fatty acid β-oxidation ACADM deficiency dehydrogenase, C-4 to C-12 straight chain Lpin2 -

Targeted Disruption of Shp2 in Chondrocytes Leads to Metachondromatosis with Multiple Cartilaginous Protrusions
ORIGINAL ARTICLE JBMR Targeted Disruption of Shp2 in Chondrocytes Leads to Metachondromatosis With Multiple Cartilaginous Protrusions Harry KW Kim,1,2 Gen‐Sheng Feng,3 Di Chen,4 Philip D King,5 and Nobuhiro Kamiya1,2 1Texas Scottish Rite Hospital for Children, Dallas, TX, USA 2Orthopaedic Surgery, University of Texas Southwestern Medical Center, Dallas, TX, USA 3Department of Pathology and Division of Biological Sciences, University of California San Diego, La Jolla, CA, USA 4Department of Biochemistry, Rush University Medical Center, Chicago, IL, USA 5Department of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI, USA ABSTRACT Metachondromatosis is a benign bone disease predominantly observed in the hands and feet of children or young adults demonstrating two different manifestations: a cartilage‐capped bony outgrowth on the surface of the bone called exostosis and ectopic cartilaginous nodules inside the bone called enchondroma. Recently, it has been reported that loss‐of‐function mutations of the SHP2 gene, which encodes the SHP2 protein tyrosine phosphatase, are associated with metachondromatosis. The purpose of this study was to investigate the role of SHP2 in postnatal cartilage development, which is largely unknown. We disrupted Shp2 during the postnatal stage of mouse development in a chondrocyte‐specific manner using a tamoxifen‐inducible system. We found tumor‐like nodules on the hands and feet within a month after the initial induction. The SHP2‐deficient mice demonstrated an exostosis‐like and enchondroma‐like phenotype in multiple bones of the hands, feet, and ribs as assessed by X‐ray and micro‐computed tomography (CT). Histological assessment revealed the disorganization of the growth plate cartilage, a cartilaginous protrusion from the epiphyseal bone, and ectopic cartilage nodules within the bones, which is consistent with the pathological features of metachondromatosis in humans (ie, both exostosis and enchondroma). -

WES Gene Package Multiple Congenital Anomalie.Xlsx
Whole Exome Sequencing Gene package Multiple congenital anomalie, version 5, 1‐2‐2018 Technical information DNA was enriched using Agilent SureSelect Clinical Research Exome V2 capture and paired‐end sequenced on the Illumina platform (outsourced). The aim is to obtain 8.1 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA‐MEM algorithm (reference: http://bio‐bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x A4GALT [Blood group, P1Pk system, P(2) phenotype], 111400 607922 101 100 100 99 [Blood group, P1Pk system, p phenotype], 111400 NOR polyagglutination syndrome, 111400 AAAS Achalasia‐addisonianism‐alacrimia syndrome, 231550 605378 73 100 100 100 AAGAB Keratoderma, palmoplantar,