Complex Hereditary Spastic Paraplegia Associated with Episodic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tbamitchodral L Alizaion of the 4Aminobutyrate-2-&Oxoglutarate
5d.em. J. (lWg77) 161,9O.-307 3O1 Printed in Great Britain Tbamitchodral L alizaion of the 4Aminobutyrate-2-&Oxoglutarate Transminase from Ox Brait By INGER SCHOUSDOE,* BIRGIT 1MO* and ARNE SCHOUSBOEt Department ofBDahemistry At andC*, University ofCopenhagen, 2200 Copenhagen M, Denark (Receved 4 June 1976) In order to determine the intramitochondrial location of 4-aminobutyrate transaminase, mitochondria were prepared from ox brain and freed from myelin and syiaptosomes by using conventional demitygradient-centrifugation techniques, and the purity was checked electron-microscopically. Iner and outer mimbrenes and matrix were prepared from the mitochondria by large-amplitude sweling and subsequent density-gradient centrfugationt The fractions were characterized by using both electron microscopy and differnt marker enzymes. From the specific activity of the 4-aminobutyrate transaminase in the submitochondrial fractions it was concluded that this enzyme is associated with the inner mitochondrial membrane. It is generally agreed that the 4-aminobutyrate-2- pyridoxal phosphate were from Sigma Chemical oxoglutarate transaminase (EC2.6.1.19) from brain is Co., St. Louis, MO, U.S.A. Ficoll was from mainly associated with free mitochondria (Salganicoff Pharmacia, Uppsala, Sweden, and crystallized & De Robertis, 1963, 1965; van den Berget al., 1965; bovine serum albumin was from BDH Biochemicals, van Kempen et at., 1965; Balazs et al., 1966; Poole, Dorset, U.K. 4-Amino[1-'4C]butyrate (sp. Waksman et al., 1968; Reijnierse et al., 1975), radioactivity 50mCi/mmol) and [1-14qtyramine (sp. and a preparation of a crude mitochondrial fraction radioactivity 9mCi/mmol) were obtained from was used by Schousboe et al. (1973) and Maitre et al. -

Alternative Acetate Production Pathways in Chlamydomonas Reinhardtii During Dark Anoxia and the Dominant Role of Chloroplasts in Fermentative Acetate Productionw
This article is a Plant Cell Advance Online Publication. The date of its first appearance online is the official date of publication. The article has been edited and the authors have corrected proofs, but minor changes could be made before the final version is published. Posting this version online reduces the time to publication by several weeks. Alternative Acetate Production Pathways in Chlamydomonas reinhardtii during Dark Anoxia and the Dominant Role of Chloroplasts in Fermentative Acetate ProductionW Wenqiang Yang,a,1 Claudia Catalanotti,a Sarah D’Adamo,b Tyler M. Wittkopp,a,c Cheryl J. Ingram-Smith,d Luke Mackinder,a Tarryn E. Miller,b Adam L. Heuberger,e Graham Peers,f Kerry S. Smith,d Martin C. Jonikas,a Arthur R. Grossman,a and Matthew C. Posewitzb a Carnegie Institution for Science, Department of Plant Biology, Stanford, California 94305 b Colorado School of Mines, Department of Chemistry and Geochemistry, Golden, Colorado 80401 c Stanford University, Department of Biology, Stanford, California 94305 d Clemson University, Department of Genetics and Biochemistry, Clemson, South Carolina 29634 e Colorado State University, Proteomics and Metabolomics Facility, Fort Collins, Colorado 80523 f Colorado State University, Department of Biology, Fort Collins, Colorado 80523 ORCID ID: 0000-0001-5600-4076 (W.Y.) Chlamydomonas reinhardtii insertion mutants disrupted for genes encoding acetate kinases (EC 2.7.2.1) (ACK1 and ACK2) and a phosphate acetyltransferase (EC 2.3.1.8) (PAT2, but not PAT1) were isolated to characterize fermentative acetate production. ACK1 and PAT2 were localized to chloroplasts, while ACK2 and PAT1 were shown to be in mitochondria. -

Mutation of the Fumarase Gene in Two Siblings with Progressive Encephalopathy and Fumarase Deficiency T
Mutation of the Fumarase Gene in Two Siblings with Progressive Encephalopathy and Fumarase Deficiency T. Bourgeron,* D. Chretien,* J. Poggi-Bach, S. Doonan,' D. Rabier,* P. Letouze,I A. Munnich,* A. R6tig,* P. Landneu,* and P. Rustin* *Unite de Recherches sur les Handicaps Genetiques de l'Enfant, INSERM U393, Departement de Pediatrie et Departement de Biochimie, H6pital des Enfants-Malades, 149, rue de Sevres, 75743 Paris Cedex 15, France; tDepartement de Pediatrie, Service de Neurologie et Laboratoire de Biochimie, Hopital du Kremlin-Bicetre, France; IFaculty ofScience, University ofEast-London, UK; and IService de Pediatrie, Hopital de Dreux, France Abstract chondrial enzyme (7). Human tissue fumarase is almost We report an inborn error of the tricarboxylic acid cycle, fu- equally distributed between the mitochondria, where the en- marase deficiency, in two siblings born to first cousin parents. zyme catalyzes the reversible hydration of fumarate to malate They presented with progressive encephalopathy, dystonia, as a part ofthe tricarboxylic acid cycle, and the cytosol, where it leucopenia, and neutropenia. Elevation oflactate in the cerebro- is involved in the metabolism of the fumarate released by the spinal fluid and high fumarate excretion in the urine led us to urea cycle. The two isoenzymes have quite homologous struc- investigate the activities of the respiratory chain and of the tures. In rat liver, they differ only by the acetylation of the Krebs cycle, and to finally identify fumarase deficiency in these NH2-terminal amino acid of the cytosolic form (8). In all spe- two children. The deficiency was profound and present in all cies investigated so far, the two isoenzymes have been found to tissues investigated, affecting the cytosolic and the mitochon- be encoded by a single gene (9,10). -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Anti-Inflammatory Role of Curcumin in LPS Treated A549 Cells at Global Proteome Level and on Mycobacterial Infection
Anti-inflammatory Role of Curcumin in LPS Treated A549 cells at Global Proteome level and on Mycobacterial infection. Suchita Singh1,+, Rakesh Arya2,3,+, Rhishikesh R Bargaje1, Mrinal Kumar Das2,4, Subia Akram2, Hossain Md. Faruquee2,5, Rajendra Kumar Behera3, Ranjan Kumar Nanda2,*, Anurag Agrawal1 1Center of Excellence for Translational Research in Asthma and Lung Disease, CSIR- Institute of Genomics and Integrative Biology, New Delhi, 110025, India. 2Translational Health Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, 110067, India. 3School of Life Sciences, Sambalpur University, Jyoti Vihar, Sambalpur, Orissa, 768019, India. 4Department of Respiratory Sciences, #211, Maurice Shock Building, University of Leicester, LE1 9HN 5Department of Biotechnology and Genetic Engineering, Islamic University, Kushtia- 7003, Bangladesh. +Contributed equally for this work. S-1 70 G1 S 60 G2/M 50 40 30 % of cells 20 10 0 CURI LPSI LPSCUR Figure S1: Effect of curcumin and/or LPS treatment on A549 cell viability A549 cells were treated with curcumin (10 µM) and/or LPS or 1 µg/ml for the indicated times and after fixation were stained with propidium iodide and Annexin V-FITC. The DNA contents were determined by flow cytometry to calculate percentage of cells present in each phase of the cell cycle (G1, S and G2/M) using Flowing analysis software. S-2 Figure S2: Total proteins identified in all the three experiments and their distribution betwee curcumin and/or LPS treated conditions. The proteins showing differential expressions (log2 fold change≥2) in these experiments were presented in the venn diagram and certain number of proteins are common in all three experiments. -

Citric Acid Cycle
CHEM464 / Medh, J.D. The Citric Acid Cycle Citric Acid Cycle: Central Role in Catabolism • Stage II of catabolism involves the conversion of carbohydrates, fats and aminoacids into acetylCoA • In aerobic organisms, citric acid cycle makes up the final stage of catabolism when acetyl CoA is completely oxidized to CO2. • Also called Krebs cycle or tricarboxylic acid (TCA) cycle. • It is a central integrative pathway that harvests chemical energy from biological fuel in the form of electrons in NADH and FADH2 (oxidation is loss of electrons). • NADH and FADH2 transfer electrons via the electron transport chain to final electron acceptor, O2, to form H2O. Entry of Pyruvate into the TCA cycle • Pyruvate is formed in the cytosol as a product of glycolysis • For entry into the TCA cycle, it has to be converted to Acetyl CoA. • Oxidation of pyruvate to acetyl CoA is catalyzed by the pyruvate dehydrogenase complex in the mitochondria • Mitochondria consist of inner and outer membranes and the matrix • Enzymes of the PDH complex and the TCA cycle (except succinate dehydrogenase) are in the matrix • Pyruvate translocase is an antiporter present in the inner mitochondrial membrane that allows entry of a molecule of pyruvate in exchange for a hydroxide ion. 1 CHEM464 / Medh, J.D. The Citric Acid Cycle The Pyruvate Dehydrogenase (PDH) complex • The PDH complex consists of 3 enzymes. They are: pyruvate dehydrogenase (E1), Dihydrolipoyl transacetylase (E2) and dihydrolipoyl dehydrogenase (E3). • It has 5 cofactors: CoASH, NAD+, lipoamide, TPP and FAD. CoASH and NAD+ participate stoichiometrically in the reaction, the other 3 cofactors have catalytic functions. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -
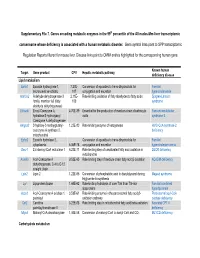
Supplementary File 7. Genes Encoding Metabolic Enzymes in The
Supplementary File 7. Genes encoding metabolic enzymes in the 99th percentile of the All nodes-Mm-liver transcriptomic consensome whose deficiency is associated with a human metabolic disorder. Gene symbol links point to SPP transcriptomic Regulation Reports filtered for mouse liver. Disease links point to OMIM entries highlighted for the corresponding human gene. Known human Target Gene product CPV Hepatic metabolic pathway deficiency disease Lipid metabolism Ephx1 Epoxide hydroxylase 1, 7.27E- Conversion of epoxides to trans-dihydrodiols for Familial microsomal xenobiotic 107 conjugation and excretion hypercholanemia Aldh3a2 Aldehyde dehydrogenase 3 2.11E- Rate-limiting oxidation of fatty aldehydes to fatty acids Sjogren-Larsson family, member A2 (fatty 103 syndrome aldehyde dehydrogenase) Ehhadh Enoyl-Coenzyme A, 4.70E-89 Essential for the production of medium-chain dicarboxylic Fanconi renotubular hydratase/3-hydroxyacyl acids syndrome 3 Coenzyme A dehydrogenase Hmgcs2 3-hydroxy-3-methylglutaryl- 1.23E-80 Rate-limiting enzyme of ketogenesis HMG-CoA synthase-2 coenzyme A synthase 2, deficiency mitochondrial Ephx2 Epoxide hydrolase 2, Conversion of epoxides to trans-dihydrodiols for Familial cytoplasmic 6.06E-78 conjugation and excretion hypercholesterolemia Decr1 2,4-dienoyl-CoA reductase 1 6.23E-71 Rate-limiting step of unsaturated fatty acid oxidation in DECR deficiency mitochondria Acadm Acyl-Coenzyme A 9.52E-65 Rate limiting step of medium-chain fatty acid β-oxidation ACADM deficiency dehydrogenase, C-4 to C-12 straight chain Lpin2 -

Microrna–Target Pairs in the Rat Kidney Identified by Microrna Microarray, Proteomic, and Bioinformatic Analysis Zhongmin Tian,1,2 Andrew S
Downloaded from genome.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Letter MicroRNA–target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformatic analysis Zhongmin Tian,1,2 Andrew S. Greene,1,2 Jennifer L. Pietrusz,1 Isaac R. Matus,2 and Mingyu Liang1,3 1Department of Physiology, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, USA; 2Biotechnology and Biomedical Engineering Center, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, USA Mammalian genomes contain several hundred highly conserved genes encoding microRNAs. In silico analysis has predicted that a typical microRNA may regulate the expression of hundreds of target genes, suggesting miRNAs might have broad biological significance. A major challenge is to obtain experimental evidence for predicted microRNA–target pairs. We reasoned that reciprocal expression of a microRNA and a predicted target within a physiological context would support the presence and relevance of a microRNA–target pair. We used microRNA microarray and proteomic techniques to analyze the cortex and the medulla of rat kidneys. Of the 377 microRNAs analyzed, we identified 6 as enriched in the renal cortex and 11 in the renal medulla. From ∼2100 detectable protein spots in two-dimensional gels, we identified 58 proteins as more abundant in the renal cortex and 72 in the renal medulla. The differential expression of several microRNAs and proteins was verified by real-time PCR and Western blot analyses, respectively. Several pairs of reciprocally expressed microRNAs and proteins were predicted to be microRNA–target pairs by TargetScan, PicTar, or miRanda. Seven pairs were predicted by two algorithms and two pairs by all three algorithms. -
![(ACO2) Mouse Monoclonal Antibody [Clone ID: OTI7G4] Product Data](https://docslib.b-cdn.net/cover/3055/aco2-mouse-monoclonal-antibody-clone-id-oti7g4-product-data-823055.webp)
(ACO2) Mouse Monoclonal Antibody [Clone ID: OTI7G4] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA500873 Aconitase 2 (ACO2) Mouse Monoclonal Antibody [Clone ID: OTI7G4] Product data: Product Type: Primary Antibodies Clone Name: OTI7G4 Applications: FC, IF, WB Recommended Dilution: WB 1:2000, IF 1:100, Flow 1:100 Reactivity: Human, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human ACO2 (NP_001089) produced in HEK293T cell. Formulation: PBS (pH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 85.4 kDa Gene Name: aconitase 2 Database Link: NP_001089 Entrez Gene 11429 MouseEntrez Gene 79250 RatEntrez Gene 50 Human Q99798 Background: The protein encoded by this gene belongs to the aconitase/IPM isomerase family. It is an enzyme that catalyzes the interconversion of citrate to isocitrate via cis-aconitate in the second step of the TCA cycle. This protein is encoded in the nucleus and functions in the mitochondrion. It was found to be one of the mitochondrial matrix proteins that are preferentially degraded by the serine protease 15(PRSS15), also known as Lon protease, after oxidative modification. This product is to be used for laboratory only. Not for diagnostic or therapeutic use. -

Hepatic Cystathionine Β-Synthase Activity Is Increased by Greater Postruminal Supply of Met During the Periparturient Period in Dairy Cows
ORIGINAL RESEARCH CURRENT DEVELOPMENTS IN NUTRITION Nutritional Biochemistry and Physiologic Mechanisms Hepatic Cystathionine β-Synthase Activity Is Increased by Greater Postruminal Supply of Met during the Periparturient Period in Dairy Cows Mario Vailati-Riboni,1 Fernanda Batistel,1 Rainie RCS Yambao,1 Claudia Parys,2 Yuan-Xiang Pan,3 and Juan J Loor1 1Department of Animal Sciences and Division of Nutritional Sciences, University of Illinois, Urbana, IL, USA; 2Evonik Nutrition & Care GmbH, Hanau-Wolfgang, Germany; and 3Department of Food Science and Human Nutrition and Division of Nutritional Sciences, University of Illinois, Urbana, IL, USA ABSTRACT Background: Postruminal supply of Met during the periparturient period enhances production efficiency (feed conversion to milk) in dairy cows partly through alleviation of oxidant and inflammatory status. Whether alterations in hepatic 1-carbon metabolism (major contributor of antioxidants) and/or energy metabolism contribute to these beneficial effects is unknown. Objectives: To investigate alterations in hepatic 1-carbon and energy metabolism and associations with plasma amino acids (AAs) and production efficiency in response to enhanced postruminal supply of Met. Methods: Holstein cows (n = 30 per group) were fed during the last 28 d of pregnancy a control diet (CON) or the control plus ethylcellulose rumen-protected Met (MET; 0.9 g/kg of dry matter intake). Plasma (n = 15 per group) and liver tissue (n = 10 per group) were collected throughout the periparturient period to evaluate AA profiles, activity of the tricarboxylic acid cycle, and 1-carbon metabolism via mRNA abundance, enzyme activity, and targeted metabolomics. Results: Cows in the MET group had greater overall (27%, P = 0.027) plasma Met concentrations, but had similar total plasma AA concentrations. -

Whole Exome Sequencing Gene Package Intellectual Disability, Version 9.1, 31-1-2020
Whole Exome Sequencing Gene package Intellectual disability, version 9.1, 31-1-2020 Technical information DNA was enriched using Agilent SureSelect DNA + SureSelect OneSeq 300kb CNV Backbone + Human All Exon V7 capture and paired-end sequenced on the Illumina platform (outsourced). The aim is to obtain 10 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate and non-unique reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA-MEM algorithm (reference: http://bio-bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x A2ML1 {Otitis media, susceptibility to}, 166760 610627 66 100 100 96 AARS1 Charcot-Marie-Tooth disease, axonal, type 2N, 613287 601065 63 100 97 90 Epileptic encephalopathy, early infantile, 29, 616339 AASS Hyperlysinemia,