Exopeptidase Digestion in Combination with Field Desorption Mass Spectrometry for Amino Acid Sequence Determination
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

JASON MARC GOLDSTEIN the Isolation, Characterization
JASON MARC GOLDSTEIN The Isolation, Characterization and Cloning of Three Novel Peptidases From Streptoccocus gordonii: Their Potential Roles in Subacute Bacterial Endocarditis (Under the Direction of JAMES TRAVIS) Streptococcus gordonii is generally considered a benign inhabitant of the oral microflora yet is a primary etiological agent in the development of subacute bacterial endocarditis (SBE), an inflammatory state that propagates thrombus formation and tissue damage on the surface of heart valves. Colonization and adherence mechanisms have been identified, yet factors necessary to sustain growth remain unidentified. Strain FSS2 produced three extracellular aminopeptidase activities during growth in neutral pH- controlled batch cultures. The first included a serine-class dipeptidyl-aminopeptidase, an x-Pro DPP (Sg-xPDPP) found as an 85 kDa monomer by SDS-PAGE while appearing as a homodimer under native conditions. Kinetic studies indicated a unique and stringent x- Pro specificity comparable to the DPPIV/CD26 and lactococcal x-Pro DPP families. Isolation of the full-length gene uncovered a 759-amino acid polypeptide with a mass of 87,115 Da and theoretical pI of 5.6. Significant homology was found with PepX gene family members from Lactobacillus ssp. and Lactococcus ssp., and putative streptococcal x-Pro DPPs. The second activity was a putative serine-class arginine aminopeptidase (Sg- RAP) with some cysteine-class characteristics. It was found as a protein monomer of 70 kDa under denaturing conditions. Nested PCR cloning enabled the isolation of a 324 bp- long DNA fragment encoding the protein’s 108 amino acid N-terminus. Culture activity profiles and N-terminal sequence analysis indicated the release of this protein from the cell surface. -

High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma
molecules Article High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma Elisa Maffioli 1,2 , Zhenze Jiang 3, Simona Nonnis 1,2 , Armando Negri 1,2, Valentina Romeo 1, Christopher B. Lietz 3, Vivian Hook 3,4, Giuseppe Ristagno 5, Giuseppe Baselli 6, Erik B. Kistler 7,8 , Federico Aletti 9, Anthony J. O’Donoghue 3,* and Gabriella Tedeschi 1,2,* 1 Department of Veterinary Medicine, University of Milano, 20133 Milano, Italy; elisa.maffi[email protected] (E.M.); [email protected] (S.N.); [email protected] (A.N.); [email protected] (V.R.) 2 Centre for Nanostructured Materials and Interfaces (CIMAINA), University of Milano, 20133 Milano, Italy 3 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093, USA; [email protected] (Z.J.); [email protected] (C.B.L.); [email protected] (V.H.) 4 Department of Neurosciences, School of Medicine, University of California San Diego, La Jolla, CA 92093, USA 5 Department of Pathophysiology and Transplantation, University of Milan, 20133 Milan, Italy; [email protected] 6 Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano, 20133 Milan, Italy; [email protected] 7 Department of Anesthesiology & Critical Care, University of California San Diego, La Jolla, CA 92093, USA; [email protected] 8 Department of Anesthesiology & Critical Care, VA San Diego HealthCare System, San Diego, CA 92161, USA 9 Department of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA; [email protected] * Correspondence: [email protected] (A.J.O.); [email protected] (G.T.); Tel.: +1-8585345360 (A.J.O.); +39-02-50318127 (G.T.) Academic Editor: Paolo Iadarola Received: 28 July 2020; Accepted: 4 September 2020; Published: 6 September 2020 Abstract: Proteomic technologies have identified 234 peptidases in plasma but little quantitative information about the proteolytic activity has been uncovered. -
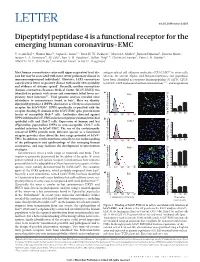
Dipeptidyl Peptidase 4 Is a Functional Receptor for the Emerging Human Coronavirus-EMC
LETTER doi:10.1038/nature12005 Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC V. Stalin Raj1*, Huihui Mou2*, Saskia L. Smits1,3, Dick H. W. Dekkers4, Marcel A. Mu¨ller5, Ronald Dijkman6, Doreen Muth5, Jeroen A. A. Demmers4, Ali Zaki7, Ron A. M. Fouchier1, Volker Thiel6,8, Christian Drosten5, Peter J. M. Rottier2, Albert D. M. E. Osterhaus1, Berend Jan Bosch2 & Bart L. Haagmans1 Most human coronaviruses cause mild upper respiratory tract dis- antigen-related cell adhesion molecules (CEACAM)10 to enter cells, ease but may be associated with more severe pulmonary disease in whereas for several Alpha- and Betacoronaviruses, two peptidases immunocompromised individuals1. However, SARS coronavirus have been identified as receptors (aminopeptidase N (APN, CD13) caused severe lower respiratory disease with nearly 10% mortality for hCoV-229E and several animal coronaviruses11,12, and angiotensin and evidence of systemic spread2. Recently, another coronavirus (human coronavirus-Erasmus Medical Center (hCoV-EMC)) was a identified in patients with severe and sometimes lethal lower res- )] Vero –1 piratory tract infection3,4. Viral genome analysis revealed close 9 ml 8 5 50 relatedness to coronaviruses found in bats . Here we identify 7 dipeptidyl peptidase 4 (DPP4; also known as CD26) as a functional 6 5 receptor for hCoV-EMC. DPP4 specifically co-purified with the 4 receptor-binding S1 domain of the hCoV-EMC spike protein from 3 Relative cell number log[GE (TCID 0 20 40 lysates of susceptible Huh-7 cells. Antibodies directed against 0 101 102 103 104 DPP4 inhibited hCoV-EMC infection of primary human bronchial b )] COS-7 –1 epithelial cells and Huh-7 cells. -

And Exopeptidases in a Processing Enzyme System: Activation
Proc. Nail. Acad. Sci. USA Vol. 85, pp. 5468-5472, August 1988 Biochemistry Relationship between endo- and exopeptidases in a processing enzyme system: Activation of an endoprotease by the aminopeptidase B-like activity in somatostatin-28 convertase (brain cortex/basic amino acid pairs/peptide substrates/protease inhibitors/prohormone maturation) SOPHIE GOMEZ, PABLO GLUSCHANKOF, AGNES LEPAGE, AND PAUL COHEN Groupe de Neurobiochimie Cellulaire et Moldculaire, Universitd Pierre et Marie Curie, Unitd Associde 554 au Centre National de la Recherche Scientifique, 96 boulevard Raspail, 75006 Paris, France Communicated by I. Robert Lehman, April 8, 1988 (receivedfor review December 15, 1987) ABSTRACT The somatostatin-28 convertase activity in- somatostatin-14 and the amino-terminal dodecapeptide so- volved in vitro in the processing of somatostatin-28 into the matostatin-28-(1-12) (16). neuropeptides somatostatin-28-(1-12) and somatostatin-14 is We have described an endoprotease that cleaves the composed of an endoprotease and a basic aminopeptidase. We peptide bond on the amino side ofthe Arg-Lys doublet in the report herein on the purification to apparent homogeneity of somatostatin-28 sequence (17, 18), releasing the somato- these two constituents and on their functional interrelationship. statin-28-(1-12) fragment and [Arg-2,Lys-1]somatostatin-14. In particular we observed that after various physicochemical The released [Arg-2,Lys-']somatostatin-14 is further proc- treatments, the 90-kDa endoprotease activity was recovered essed by an aminopeptidase B-like activity (18, 19) that is also both at this molecular mass and as a 45-kDa entity. Moreover, present in the preparation. We report herein the purification the production of [Arg2,LysJllsomatostatin-14 from somato- to apparent homogeneity ofthese two activities. -

Proteolytic Cleavage—Mechanisms, Function
Review Cite This: Chem. Rev. 2018, 118, 1137−1168 pubs.acs.org/CR Proteolytic CleavageMechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification Theo Klein,†,⊥ Ulrich Eckhard,†,§ Antoine Dufour,†,¶ Nestor Solis,† and Christopher M. Overall*,†,‡ † ‡ Life Sciences Institute, Department of Oral Biological and Medical Sciences, and Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada ABSTRACT: Proteases enzymatically hydrolyze peptide bonds in substrate proteins, resulting in a widespread, irreversible posttranslational modification of the protein’s structure and biological function. Often regarded as a mere degradative mechanism in destruction of proteins or turnover in maintaining physiological homeostasis, recent research in the field of degradomics has led to the recognition of two main yet unexpected concepts. First, that targeted, limited proteolytic cleavage events by a wide repertoire of proteases are pivotal regulators of most, if not all, physiological and pathological processes. Second, an unexpected in vivo abundance of stable cleaved proteins revealed pervasive, functionally relevant protein processing in normal and diseased tissuefrom 40 to 70% of proteins also occur in vivo as distinct stable proteoforms with undocumented N- or C- termini, meaning these proteoforms are stable functional cleavage products, most with unknown functional implications. In this Review, we discuss the structural biology aspects and mechanisms -
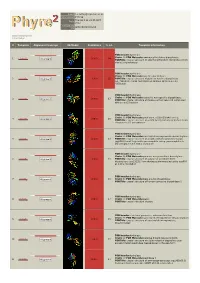
Phyre 2 Results for P15288
Email [email protected] Description P15288 Thu Jan 5 11:34:45 GMT Date 2012 Unique Job d2087d8d303c52c9 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:hydrolase Chain: B: PDB Molecule:aminoacyl-histidine dipeptidase; 1 c3mruB_ 100.0 64 Alignment PDBTitle: crystal structure of aminoacylhistidine dipeptidase from vibrio2 alginolyticus PDB header:hydrolase Chain: B: PDB Molecule:xaa-his dipeptidase; 2 c2qyvB_ Alignment 100.0 55 PDBTitle: crystal structure of putative xaa-his dipeptidase (yp_718209.1) from2 haemophilus somnus 129pt at 2.11 a resolution PDB header:hydrolase Chain: A: PDB Molecule:cytosolic non-specific dipeptidase; 3 c2zogA_ 100.0 17 Alignment PDBTitle: crystal structure of mouse carnosinase cn2 complexed with zn and2 bestatin PDB header:hydrolase Chain: B: PDB Molecule:peptidase, m20/m25/m40 family; 4 c2pokB_ 100.0 16 Alignment PDBTitle: crystal structure of a m20 family metallo peptidase from streptococcus2 pneumoniae PDB header:hydrolase Chain: A: PDB Molecule:succinyl-diaminopimelate desuccinylase; 5 c3pfeA_ Alignment 100.0 13 PDBTitle: crystal structure of a m20a metallo peptidase (dape, lpg0809) from2 legionella pneumophila subsp. pneumophila str. philadelphia 1 at 1.503 a resolution PDB header:hydrolase Chain: B: PDB Molecule:putative acetylornithine deacetylase; 6 c3pfoB_ Alignment 100.0 16 PDBTitle: crystal structure of a putative acetylornithine deacetylase (rpa2325)2 from rhodopseudomonas palustris cga009 at 1.90 a resolution PDB -

Contribution of Endo- and Exopeptidases to Formation of Non-Protein N During Ensiling of Alfalfa
Contribution of endo- and exopeptidases to formation of non-protein N during ensiling of alfalfa Dr. Xusheng Guo,1 W. Cheng,1 L. Tao,2 Yu Zhu,2 F.Y. Yang 2 & H. Zhou2 1.School of Life Science, Lanzhou University 1. International Centre for Tibetan Plateau Ecosystem Management (ICTPEM), Lanzhou University 2. Institute of Grassland Science, China Agricultural University Hämeenlinna, Finland 2-4 July 2012 Introduction Alfalfa (Medicago Sativa L.) is well known for its high nutritive value However, after ensiling: N use efficiency Extensive ppyroteolysis Reduce True protein NPN(Peptide, FAA, NH3-N etc.) Silage DM intake (44-87% of Total N; Muck, 1987) Silage Fermentation Proteolysis in ensiled forage mainly results from plant proteases (Ohshima and McDonald, 1978; McKersie, 1981; Heron et al., 1988). Proteases (peptidases) are divided into 2 classes (NC-IUBMB, 1992): Exopeptidase Endopeptidase Objectives Proteases (peptidases, E.C.3.4) Endopeptidases Exopeptidases ?! Ser ine pep tidase (E .C .3 .4 .21) Aminopeptidase (EC 3.4.11) Carboxypeptidase (EC 3.4.16) Cysteine peptidase (E.C.3.4.22) Dipeptidase (EC 3.4.13) Aspartic Peptidase (E.C.3.4.23) Dipeptidyl-peptidase (EC 3.4.14) Metallopeptidase (E.C.3.4.24) Tripeptidyl-peptidase (EC 3.4.14) Peptidyl-dipeptidase (EC 3.4.15) Aims of our research were: 1. To clarify the classes of exo- and endopeptidases that are involved in proteolysis within ensiled alfalfa. 2. To determine the contribution of these peptidases to the formation of different NPN compounds (peptide-N, FAA-N, and NH3-N) during -

Cathepsin B Carboxydipeptidase Specificity Analysis Using Internally
Biochem. J. (2002) 368, 365–369 (Printed in Great Britain) 365 Cathepsin B carboxydipeptidase specificity analysis using internally quenched fluorescent peptides Maria Helena S. CEZARI, Luciano PUZER, Maria Aparecida JULIANO, Adriana K. CARMONA and Luiz JULIANO1 Department of Biophysics, Escola Paulista de Medicina, Universidade Federal de Sa4 o Paulo, Rua Tre# s de Maio, 100, Sa4 o Paulo 04044-020, Brazil We have examined in detail the specificity of the subsites S",S#, analysis of its specificity. The subsite S" accepted preferentially h h S" and S# for the carboxydipeptidase activity of cathepsin B by basic amino acids for hydrolysis; however, substrates with synthesizing and assaying four series of internally quenched phenylalanine and aliphatic side-chain-containing amino acids at #%& fluorescent peptides based on the sequence Dnp-GFRFW-OH, P" had lower Km values. Despite the presence of Glu at S#, where Dnp (2,4-dinitrophenyl) is the quenching group of the this subsite presented clear preference for aromatic amino acid fluorescence of the tryptophan residue. Each position, except the residues, and the substrate with a lysine residue at P# was h glycine, was substituted with 15 different naturally occurring hydrolysed better than that containing an arginine residue. S" is h amino acids. Based on the results we obtained, we also synthesized essentially a hydrophobic subsite, and S# has particular efficient and sensitive substrates that contained o-aminobenzoic preference for phenylalanine or tryptophan residues. acid and 3-Dnp-(2,3-diaminopropionic acid), or ε-amino-Dnp- Lys, as the fluorescence donor–receptor pair. The higher kinetic parameter values for the carboxydipeptidase compared with the Key words: fluorescent peptide, fluorogenic substrate, proteinase, endopeptidase activity of cathepsin B allowed an accurate thiol protease. -

Tagzyme™ Handbook
TAGZyme ™ Handbook For Exoproteolytic cleavage of N-terminal His tags July 2015 © 2001–2015 QIAGEN, all rights reserved. www.qiagen.com Contents Kit Contents 5 Storage and Stability 6 Safety Information 6 Product Use Limitations 7 Product Warranty and Satisfaction Guarantee 7 Technical Assistance 7 Introduction 8 The TAGZyme Principle 9 Proteins with intrinsic DAPase stop points 9 Proteins without intrinsic DAPase stop points 10 Applications for the TAGZyme System 12 TAGZyme pQE Vectors 13 QIA express pQE vectors 13 Regulation of expression – pREP4 plasmid 14 E. coli host strains 15 The Cloning Procedure 17 Cleavage Protocols 20 Notes Before Starting 20 His tag design 20 Expression, Ni-NTA purification, and desalting of His-tagged proteins 20 Buffers and enzyme activity 21 DAPase Enzyme 21 Qcyclase Enzyme 21 pGAPase Enzyme 22 Cysteamine activation 22 Stepwise Use of the TAGZyme System 22 Analysis during the TAGZyme procedure 23 Cleavage of N-terminal 6xHis tags using DAPase Enzyme 25 Buffer preparation 25 Desalting 25 Protocol 1. DAPase digestion (small-scale pilot experiment) for proteins containing intrinsic DAPase stop points 26 TAGZyme Handbook 07/2015 3 Protocol 2. Preparative DAPase digestion for proteins containing intrinsic DAPase stop points 28 Protocol 3. Removal of DAPase Enzyme by subtractive IMAC 29 Cleavage of N-terminal His tags using DAPase, Qcyclase, and pGAPase Enzymes 30 Buffer preparation 30 Desalting 30 Protocol 4. DAPase – Qcyclase digestion (small-scale pilot experiment) 31 Protocol 5. Preparative DAPase – Qcyclase digestion 33 Protocol 6. Removal of DAPase and Qcyclase Enzymes by subtractive IMAC 34 Protocol 7. pGAPase Digestion 35 Protocol 8. -

Structures and Mechanism of Dipeptidyl Peptidases 8 and 9
Structures and mechanism of dipeptidyl peptidases PNAS PLUS 8 and 9, important players in cellular homeostasis and cancer Breyan Rossa,b,1, Stephan Krappb, Martin Augustinb, Reiner Kierfersauerb, Marcelino Arciniegac, Ruth Geiss-Friedlanderd, and Robert Hubera,e,f,1 aMax Planck Institut für Biochemie, D-82152 Martinsried, Germany; bProteros Biostructures GmbH, D-82152 Martinsried, Germany; cDepartment of Biochemistry and Structural Biology, Institute of Cellular Physiology, Universidad Nacional Autónoma de México, 04510 Mexico City, Mexico; dAbteilung für Molekularbiologie, Universitätsmedizin Göttingen, D-37073 Göttingen, Germany; eZentrum für Medizinische Biotechnologie, Universität Duisburg-Essen, D-45117 Essen, Germany; and fFakultät für Chemie, Technische Universität München, D-85747 Garching, Germany Contributed by Robert Huber, December 12, 2017 (sent for review October 16, 2017; reviewed by Ingrid De Meester and Guy S. Salvesen) Dipeptidyl peptidases 8 and 9 are intracellular N-terminal dipeptidyl be processed by DPP9, thereby regulating B cell signaling (18). peptidases (preferentially postproline) associated with pathophysi- DPP9 activity is also connected to pathophysiological conditions, ological roles in immune response and cancer biology. While the as promoting tumoregenicity and metastasis in nonsmall cell lung DPP family member DPP4 is extensively characterized in molecular cancer (19). Recently, DPP9 fusion genes were identified in high- terms as a validated therapeutic target of type II diabetes, experimental grade serous -

Exopeptidase Catalyzed Site-Specific Bonding of Supports, Labels and Bioactive Agents to Proteins
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Biochemistry -- Faculty Publications Biochemistry, Department of 1993 EXOPEPTIDASE CATALYZED SITE-SPECIFIC BONDING OF SUPPORTS, LABELS AND BIOACTIVE AGENTS TO PROTEINS Fred W. Wagner Thomas R. Coolidge Sheldon M. Schuster Jay Stout Dwane E. Wylie See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/biochemfacpub Part of the Biochemistry Commons, Biotechnology Commons, and the Other Biochemistry, Biophysics, and Structural Biology Commons This Article is brought to you for free and open access by the Biochemistry, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Biochemistry -- Faculty Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Fred W. Wagner, Thomas R. Coolidge, Sheldon M. Schuster, Jay Stout, Dwane E. Wylie, Klaus Breddam, and William Lewis US005234820A United States Patent (19) 11 Patent Number: 5,234,820 Wagner et al. (45) Date of Patent: Aug. 10, 1993 (54) EXOPEPTDASE CATALYZED (56) References Cited SITE-SPECIFICBONDING OF SUPPORTS, LABELS AND BOACTIVE AGENTS TO FOREIGN PATENT DOCUMENTS PROTENS 0085516 1/1981 European Pat. Off. (75) Inventors: Fred W. Wagner, Walton, Nebr.; OTHER PUBLICATIONS Thomas R. Coolidge, Falls Village, Kauer, et al., The Journal of Biological Chemistry, vol. Conn.; Sheldon M. Schuster, 261, No. 23, 1986, pp. 10695-10700. Gainesville, Fla.; Jay Stout; Dwane E. Wylie, both of Lincoln, Nebr.; Primary Examiner-David M. Naff Klaus Breddan, Glostrup, Denmark; Attorney, Agent, or Firm-Merchant, Gould, Smith, William Lewis, Lincoln, Nebr. Edell, Welter & Schmidt (73) Assignees: Board of Regents of the University of 57 ABSTRACT Nebraska; BioNebraska, Inc., both of An auxiliary substance such as a label, support, or bi Lincoln, Nebr. -

Leucine Aminopeptidase: Bestatin Inhibition and a Model for Enzyme-Catalyzed Peptide Hydrolysis (X-Ray Crystallofraphy/Zinc Enzyme/Exopeptidase) STEPHEN K
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 6916-6920, August 1991 Biochemistry Leucine aminopeptidase: Bestatin inhibition and a model for enzyme-catalyzed peptide hydrolysis (x-ray crystallofraphy/zinc enzyme/exopeptidase) STEPHEN K. BURLEY*tt, PETER R. DAVID*§, AND WILLIAM N. LIPSCOMB* *Gibbs Chemical Laboratory, Harvard University, Cambridge, MA 02138; and tDepartment of Medicine, Brigham and Women's Hospital, Boston, MA 02115 Contributed by William N. Lipscomb, April 24, 1991 ABSTRACT The three-dimensional structures of native (11). In this paper, we present a detailed analysis of the bovine lens leucine aminopeptidase (EC 3.4.11.1) and its mechanism of inhibition of the enzyme by bestatin and complex with bestatin, a slow-binding inhibitor, have been discuss features of a mechanism of catalysis of peptide solved and exhaustively refined. The mode of binding of hydrolysis by LAP. This mechanism is in no way definitive, bestatin to leucine aminopeptidase may be similar to that of a but rather is intended to stimulate experimental studies ofthe tetrahedral intermediate that is thought to form during peptide biochemistry, including site-directed mutagenesis, of the bond hydrolysis. Bestatin binds in the active site with its LAPs. a-amino group and hydroxyl group coordinated to the zinc ion located in the readily exchangeable divalent cation binding site. Requirements for LAP-Catalyzed Peptide Hydrolysis Its phenylalanyl side chain is stabilized by van der Waals interactions with Met-270, Thr-359, Gly-362, Aa-451, and Hydrolytic cleavage of the amino-terminal peptide bond of a Met-454, which appear to form a terminal hydrophobic pocket. polypeptide substrate by LAP involves nucleophilic attack at The leucyl side chain binds in another hydrophobic cleft lined the carbonyl carbon atom and electrophilic attack at the by Asn-330, Ala-333, and fle-421.