Structural Characterization of Proteinaceous Rnase P from Arabidopsis Thaliana Franziska Pinker
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2010 Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis Ryan Fassnacht Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Physiology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2246 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Ryan C. Fassnacht 2010 All Rights Reserved Molecular Mechanisms Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by Ryan Christopher Fassnacht, B.S. Hampden Sydney University, 2005 M.S. Virginia Commonwealth University, 2010 Director: Valeria Mas, Ph.D., Associate Professor of Surgery and Pathology Division of Transplant Department of Surgery Virginia Commonwealth University Richmond, Virginia July 9, 2010 Acknowledgement The Author wishes to thank his family and close friends for their support. He would also like to thank the members of the molecular transplant team for their help and advice. This project would not have been possible with out the help of Dr. Valeria Mas and her endearing -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
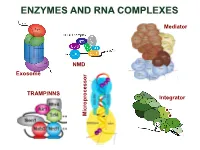
Enzymes and Rna Complexes
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

A Disease-Linked Lncrna Mutation in Rnase MRP Inhibits Ribosome Synthesis
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A disease-linked lncRNA mutation in RNase MRP inhibits ribosome synthesis Nic Roberston1, Vadim Shchepachev1, David Wright2, Tomasz W. Turowski1, Christos Spanos1, Aleksandra Helwak1, Rose Zamoyska2, David Tollervey1 1 Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, UK 2 Ashworth Laboratories, Institute of Immunology and Infection Research, University of Edinburgh, Edinburgh, UK Keywords: protein-RNA interaction; RNA-binding sites; UV crosslinking; mass spectrometry; genetic disease; Cartilage Hair Hypoplasia; ribosome synthesis; T cell activation Running title: RNase MRP defects cause ribosomopathy Highlights: • Mutations in RMRP lncRNA impair pre-rRNA processing and T cell activation • Patient derived fibroblasts show impaired pre-rRNA processing • Cells with the most common disease-linked mutation have specific processing defects • Cytoplasmic ribosomes and intact RNase MRP complexes are also reduced in these cells 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Mutations in the human RMRP gene cause Cartilage Hair Hypoplasia (CHH), an autosomal recessive disorder characterized by skeletal abnormalities and impaired T cell activation. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

A Specialized Processing Body That Is Temporally and Asymmetrically Regulated During the Cell Cycle in Saccharomyces Cerevisiae
JCB: ARTICLE A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae Tina Gill, Jason Aulds, and Mark E. Schmitt Department of Biochemistry and Molecular Biology, State University of New York Upstate Medical University, Syracuse, NY 13210 Nase mitochondrial RNA processing (MRP) is an this spot was asymmetrically found in daughter cells, essential ribonucleoprotein endoribonuclease that where the RNase MRP substrate, CLB2 mRNA, localizes. R functions in the degradation of specifi c mRNAs in- Both the mitotic exit network and fourteen early ana- volved in cell cycle regulation. We have investigated phase release pathways are nonessential but important where this processing event occurs and how it is regu- for the temporal changes in localization. Asymmetric lo- lated. As expected, results demonstrate that RNase MRP calization was found to be dependent on the locasome. is predominantly localized in the nucleolus, where it The evidence suggests that these spots are specialized processes ribosomal RNAs. However, after the initiation processing bodies for the degradation of transcripts that of mitosis, RNase MRP localizes throughout the entire are cell cycle regulated and daughter cell localized. We nucleus and in a single discrete cytoplasmic spot that have called these TAM bodies for temporal asymmetric persists until the completion of telophase. Furthermore, MRP bodies. Introduction RNase mitochondrial RNA processing (MRP) is an essential ri- 27SA preribosomal RNA at the A3 site, forming the 5.8S(s) bonucleoprotein endoribonuclease that cleaves RNA substrates ribosomal RNA (rRNA; Schmitt and Clayton, 1993; Lygerou in a site-specifi c manner and is highly conserved in eukaryotes et al., 1996). -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

Ribonuclease A
Chem. Rev. 1998, 98, 1045−1065 1045 Ribonuclease A Ronald T. Raines Departments of Biochemistry and Chemistry, University of WisconsinsMadison, Madison, Wisconsin 53706 Received October 10, 1997 (Revised Manuscript Received January 12, 1998) Contents I. Introduction 1045 II. Heterologous Production 1046 III. Structure 1046 IV. Folding and Stability 1047 A. Disulfide Bond Formation 1047 B. Prolyl Peptide Bond Isomerization 1048 V. RNA Binding 1048 A. Subsites 1048 B. Substrate Specificity 1049 C. One-Dimensional Diffusion 1049 D. Processive Catalysis 1050 VI. Substrates 1050 VII. Inhibitors 1051 Ronald T. Raines was born in 1958 in Montclair, NJ. He received Sc.B. VIII. Reaction Mechanism 1052 degrees in chemistry and biology from the Massachusetts Institute of A. His12 and His119 1053 Technology. At M.I.T., he worked with Christopher T. Walsh to reveal the reaction mechanisms of pyridoxal 5′-phosphate-dependent enzymes. B. Lys41 1054 Raines was a National Institutes of Health predoctoral fellow in the C. Asp121 1055 chemistry department at Harvard University. There, he worked with D. Gln11 1056 Jeremy R. Knowles to elucidate the reaction energetics of triosephosphate IX. Reaction Energetics 1056 isomerase. Raines was a Helen Hay Whitney postdoctoral fellow in the biochemistry and biophysics department at the University of California, A. Transphosphorylation versus Hydrolysis 1056 San Francisco. At U.C.S.F., he worked with William J. Rutter to clone, B. Rate Enhancement 1057 express, and mutate the cDNA that codes for ribonuclease A. Raines X. Ribonuclease S 1058 then joined the faculty of the biochemistry department at the University s A. S-Protein−S-Peptide Interaction 1058 of Wisconin Madison, where he is now associate professor of biochem- istry and chemistry. -

Characterization of the Mammalian RNA Exonuclease 5/NEF-Sp As a Testis-Specific Nuclear 3′′′′′ → 5′′′′′ Exoribonuclease
Downloaded from rnajournal.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Characterization of the mammalian RNA exonuclease 5/NEF-sp as a testis-specific nuclear 3′′′′′ → 5′′′′′ exoribonuclease SARA SILVA,1,2 DAVID HOMOLKA,1 and RAMESH S. PILLAI1 1Department of Molecular Biology, University of Geneva, CH-1211 Geneva 4, Switzerland 2European Molecular Biology Laboratory, Grenoble Outstation, 38042, France ABSTRACT Ribonucleases catalyze maturation of functional RNAs or mediate degradation of cellular transcripts, activities that are critical for gene expression control. Here we identify a previously uncharacterized mammalian nuclease family member NEF-sp (RNA exonuclease 5 [REXO5] or LOC81691) as a testis-specific factor. Recombinant human NEF-sp demonstrates a divalent metal ion-dependent 3′′′′′ → 5′′′′′ exoribonuclease activity. This activity is specific to single-stranded RNA substrates and is independent of their length. The presence of a 2′′′′′-O-methyl modification at the 3′′′′′ end of the RNA substrate is inhibitory. Ectopically expressed NEF-sp localizes to the nucleolar/nuclear compartment in mammalian cell cultures and this is dependent on an amino-terminal nuclear localization signal. Finally, mice lacking NEF-sp are viable and display normal fertility, likely indicating overlapping functions with other nucleases. Taken together, our study provides the first biochemical and genetic exploration of the role of the NEF-sp exoribonuclease in the mammalian genome. Keywords: NEF-sp; LOC81691; Q96IC2; REXON; RNA exonuclease 5; REXO5; 2610020H08Rik INTRODUCTION clease-mediated processing to create their final 3′ ends: poly(A) tails of most mRNAs or the hairpin structure of Spermatogenesis is the process by which sperm cells are replication-dependent histone mRNAs (Colgan and Manley produced in the male germline. -

Protein Engineering to Exploit and Explore Bovine Secretory Ribonucleases
PROTEIN ENGINEERING TO EXPLOIT A N D EXPLORE BOVINE SECRETORY RIBONUCLEASES by JIN-SOO KIM A thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biochemistry) at the UNIVERSITY OF WISCONSIN-MADISON 1994 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGEMENTS I would like to thank Dr. Ronald T. Raines for his advice and support. His scientific insight has been very helpful throughout this work. I would also like to thank the entire Raines group for their friendship and companionship. I am grateful to Dr. J. Soucek and Dr. J. Matousek for their collaboration with us, which has been a valuable part of the BS-RNase research. I thank Dr. M. Karpeisky for suggesting the protein fusion project, and Dr. G. D'Alessio and Dr. L. Mazzarella for providing the coordinates of BS-RNase. I have been generously supported by Steenbock predoctoral fellowship from the Department of Biochemistry. Finally, I thank my parents, who have encouraged (or at least not discouraged) me to pursue a career in science since I was a kid. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission ABSTRACT PROTEIN ENGINEERING TO EXPLOIT AND EXPLORE BOVINE SECRETORY RIBONUCLEASES Jin-Soo Kim Under the supervision of Dr. Ronald T. Raines at the University of Wisconsin-Madison Ribonuclease S-peptide (residues 1-20) and S-protein (residues 21- 124) are the enzymatically inactive products of the limited digestion of bovine pancreatic ribonuclease A (RNase A) by subtilisin. S-Peptide binds S-protein with high affinity to form RNase S, which has full enzymatic activity.