A Disease-Linked Lncrna Mutation in Rnase MRP Inhibits Ribosome Synthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cartilage-Hair Hypoplasia
Cartilage-hair hypoplasia Description Cartilage-hair hypoplasia is a disorder of bone growth characterized by short stature ( dwarfism) with other skeletal abnormalities; fine, sparse hair (hypotrichosis); and abnormal immune system function (immune deficiency) that can lead to recurrent infections. People with cartilage-hair hypoplasia have unusually short limbs and short stature from birth. They typically have malformations in the cartilage near the ends of the long bones in the arms and legs (metaphyseal chondrodysplasia), which then affects development of the bone itself. Most people with cartilage-hair hypoplasia are unusually flexible in some joints, but they may have difficulty extending their elbows fully. Affected individuals have hair that is lighter in color than that of other family members because the core of each hair, which contains some of the pigment that contributes the hair's color, is missing. The missing core also makes each strand of hair thinner, causing the hair to have a sparse appearance overall. Unusually light-colored skin ( hypopigmentation), malformed nails, and dental abnormalities may also be seen in this disorder. The extent of the immune deficiency in cartilage-hair hypoplasia varies from mild to severe. Affected individuals with the most severe immune problems are considered to have severe combined immunodeficiency (SCID). People with SCID lack virtually all immune protection from bacteria, viruses, and fungi and are prone to repeated and persistent infections that can be very serious or life-threatening. These infections are often caused by "opportunistic" organisms that ordinarily do not cause illness in people with a normal immune system. Most people with cartilage-hair hypoplasia, even those who have milder immune deficiency, experience infections of the respiratory system, ears, and sinuses. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
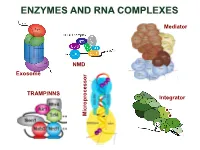
Enzymes and Rna Complexes
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

A Specialized Processing Body That Is Temporally and Asymmetrically Regulated During the Cell Cycle in Saccharomyces Cerevisiae
JCB: ARTICLE A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae Tina Gill, Jason Aulds, and Mark E. Schmitt Department of Biochemistry and Molecular Biology, State University of New York Upstate Medical University, Syracuse, NY 13210 Nase mitochondrial RNA processing (MRP) is an this spot was asymmetrically found in daughter cells, essential ribonucleoprotein endoribonuclease that where the RNase MRP substrate, CLB2 mRNA, localizes. R functions in the degradation of specifi c mRNAs in- Both the mitotic exit network and fourteen early ana- volved in cell cycle regulation. We have investigated phase release pathways are nonessential but important where this processing event occurs and how it is regu- for the temporal changes in localization. Asymmetric lo- lated. As expected, results demonstrate that RNase MRP calization was found to be dependent on the locasome. is predominantly localized in the nucleolus, where it The evidence suggests that these spots are specialized processes ribosomal RNAs. However, after the initiation processing bodies for the degradation of transcripts that of mitosis, RNase MRP localizes throughout the entire are cell cycle regulated and daughter cell localized. We nucleus and in a single discrete cytoplasmic spot that have called these TAM bodies for temporal asymmetric persists until the completion of telophase. Furthermore, MRP bodies. Introduction RNase mitochondrial RNA processing (MRP) is an essential ri- 27SA preribosomal RNA at the A3 site, forming the 5.8S(s) bonucleoprotein endoribonuclease that cleaves RNA substrates ribosomal RNA (rRNA; Schmitt and Clayton, 1993; Lygerou in a site-specifi c manner and is highly conserved in eukaryotes et al., 1996). -

The Rnase MRP and Rnase P Complexes, Will Be Summarised
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/19147 Please be advised that this information was generated on 2021-09-28 and may be subject to change. The human The human RNase MRP complex RNase MRP complex Composition, assembly and role in human disease Hans van Eenennaam Hans van Eenennaam The human RNase MRP complex Composition, assembly and role in human disease Hans van Eenennaam, 2002 The human RNase MRP complex Composition, assembly and role in human disease een wetenschappelijke proeve op het gebied van de Natuurwetenschappen, Wiskunde en Informatica PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Katholieke Universiteit Nijmegen, volgens besluit van het College van Decanen in het openbaar te verdedigen op vrijdag 14 juni 2002 des namiddags om 1:30 uur precies door Hans van Eenennaam Cover illustration Statue of the Dwarf Seneb and his family, painted limestone, height 34 cm, width 22.5 cm, geboren op 3 november 1973 Giza, Tomb of Seneb, Late Fifth - Early Sixth te Middelburg Dynasty. From: The Cairo museum Masterpieces of Egyptian Art, Francesco Tiradritti, Thames & Hudson Ltd, London, 1998 Promotor Prof. Dr. W.J. van Venrooij Co-promotor Dr. G.J.M. Pruijn voor mijn ouders Manuscriptcommissie Prof. Dr. L.B.A. van de Putte Prof. Dr. F.P.J.T. Rutjes Prof. Dr. H.F. Tabak (Universiteit van Amsterdam) ISBN 90-9015696-8 © 2002 by Hans van Eenennaam The research described in this thesis was performed at the Department of Biochemistry, Faculty of Science, University of Nijmegen, the Netherlands. -

A Novel Model for the Rnase MRP-Induced Switch Between the Different Forms of 5.8S Rrna
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 29 April 2021 doi:10.20944/preprints202104.0765.v1 Research article A novel model for the RNase MRP-induced switch between the different forms of 5.8S rRNA Xiao Li1,2,3, Janice M Zengel1 and Lasse Lindahl1 1 Department of Biological Sciences, University of Maryland Baltimore County (UMBC) 1000 Hilltop Circle, Baltimore, Maryland 21250, USA 2 Department of Biology, University of Rochester, Rochester, New York 14627, USA 3 Current address: Abbott Laboratories, San Diego, CA ribosome biogenesis; rRNA processing; RNase MRP; long/short 5.8S rRNA © 2021 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 29 April 2021 doi:10.20944/preprints202104.0765.v1 Abstract: Processing of the RNA polymerase I pre-rRNA transcript into the mature 18S, 5.8S, and 25S rRNAs requires removing the “spacer” sequences. The canonical pathway for the removal of the ITS1 spacer, located between 18S and 5.8S rRNAs in the primary transcript, involves cleavages at the 3’ end of 18S rRNA and at two sites inside ITS1. The process generates a long and a short 5.8S rRNA that differ in the number of ITS1 nucleotides retained at the 5.8S 5’ end. Here we document a novel pathway that generates the long 5.8S for ITS1 while bypassing cleavage within ITS1. It entails a single endonuclease cut at the 3’-end of 18S rRNA followed by exonuclease Xrn1 degradation of ITS1. Mutations in RNase MRP increase the accumulation of long relative to short 5.8S rRNA; traditionally this is attributed to a decreased rate of RNase MRP cleavage at its target in ITS1, called A3. -

Translation Factors and Ribosomal Proteins Control Tumor Onset and Progression: How?
Oncogene (2014) 33, 2145–2156 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW Translation factors and ribosomal proteins control tumor onset and progression: how? F Loreni1, M Mancino2,3 and S Biffo2,3 Gene expression is shaped by translational control. The modalities and the extent by which translation factors modify gene expression have revealed therapeutic scenarios. For instance, eukaryotic initiation factor (eIF)4E activity is controlled by the signaling cascade of growth factors, and drives tumorigenesis by favoring the translation of specific mRNAs. Highly specific drugs target the activity of eIF4E. Indeed, the antitumor action of mTOR complex 1 (mTORc1) blockers like rapamycin relies on their capability to inhibit eIF4E assembly into functional eIF4F complexes. eIF4E biology, from its inception to recent pharmacological targeting, is proof-of-principle that translational control is druggable. The case for eIF4E is not isolated. The translational machinery is involved in the biology of cancer through many other mechanisms. First, untranslated sequences on mRNAs as well as noncoding RNAs regulate the translational efficiency of mRNAs that are central for tumor progression. Second, other initiation factors like eIF6 show a tumorigenic potential by acting downstream of oncogenic pathways. Third, genetic alterations in components of the translational apparatus underlie an entire class of inherited syndromes known as ‘ribosomopathies’ that are associated with increased cancer risk. Taken together, data suggest that in spite of their evolutionary conservation and ubiquitous nature, variations in the activity and levels of ribosomal proteins and translation factors generate highly specific effects. Beside, as the structures and biochemical activities of several noncoding RNAs and initiation factors are known, these factors may be amenable to rational pharmacological targeting. -

Subject Index Proc
13088 Subject Index Proc. Natl. Acad. Sci. USA 91 (1994) Apoptosis in substantia nigra following developmental striatal excitotoxic Brine shrimp injury, 8117 See Artemia Visualizing hippocampal synaptic function by optical detection of Ca2l Broccol entry through the N-methyl-D-aspartate channel, 8170 See Brassica Amygdala modulation of hippocampal-dependent and caudate Bromophenacyl bromide nucleus-dependent memory processes, 8477 Bromophenacyl bromide binding to the actin-bundling protein I-plastin Distribution of corticotropin-releasing factor receptor mRNA expression inhibits inositol trisphosphate-independent increase in Ca2l in human in the rat brain and pituitary, 8777 neutrophils, 3534 Brownian dynamics Preproenkephalin promoter yields region-specific and long-term Adhesion of hard spheres under the influence of double-layer, van der expression in adult brain after direct in vivo gene transfer via a Waals, and gravitational potentials at a solid/liquid interface, 3004 defective herpes simplex viral vector, 8979 Browsers Intravenous administration of a transferrin receptor antibody-nerve Thorn-like prickles and heterophylly in Cyanea: Adaptations to extinct growth factor conjugate prevents the degeneration of cholinergic avian browsers on Hawaii?, 2810 striatal neurons in a model of Huntington disease, 9077 Bruton agammaglobulinemia Axotomy induces the expression of vasopressin receptors in cranial and Genomic organization and structure of Bruton agammaglobulinemia spinal motor nuclei in the adult rat, 9636 tyrosine kinase: Localization -

Lncrna-RMRP Promotes Proliferation, Migration and Invasion of Bladder Cancer Via Mir-206
European Review for Medical and Pharmacological Sciences 2019; 23: 1012-1021 lncRNA-RMRP promotes proliferation, migration and invasion of bladder cancer via miR-206 H.-L. CAO1, Z.-J. LIU2, P.-L. HUANG1, Y.-L. YUE3, J.-N. XI1 1Beijing Rehabilitation Hospital Affiliated to Capital Medical University, Beijing, China 2Shandong Provincial Western Hospital, Jinan, China 3Liaocheng People’s Hospital, Liaocheng, China Abstract. – OBJECTIVE: The incidence of an and American countries1. Recently, it has in- bladder cancer (BC) is common in the world, but creased its morbidity and rejuvenation. BC risk its detail mechanisms for occurrence and devel- factors are smoking and long-term exposure to opment remain unclear. Recently, long non-cod- industrial chemical products, which are related ing RNAs (lncRNAs) have been observed to play an important role in many different diseases. In to the changes in the gene level of patients; more- this research, we mainly explored the role of the over, the variety of apparent heredity is involved RNA component of mitochondrial RNA process- in the development of tumors2. The occurrence ing endoribonuclease (lncRNA-RMRP) in blad- and development of bladder cancer is a chronic der cancer. process with multi-factors and multi-steps3. Thus, MATERIALS AND METHODS: We used qRT- investigating the detail mechanisms involved in PCR to detect the expression of lncRNA-RMRP in bladder cancer patients and tumor cells, and BC bladder cancer will provide a novel strategy the clinical significance was also analyzed. The for its prevention and treatment. Long nonco- methyl thiazolyl tetrazolium (MTT) assay was ding RNAs (lncRNAs) are new members in the used to detect the cell proliferation, and we used oncology field, containing 200 nucleotide (nt)- transwell to detect the migration and invasion, long RNA molecules and stably existing in the after the lncRNA RMRP was inhibited. -

Human Ribonuclease P: Subunits, Function, and Intranuclear Localization
Downloaded from rnajournal.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (2002), 8:1–7+ Cambridge University Press+ Printed in the USA+ Copyright © 2002 RNA Society+ DOI: 10+1017+S1355838201011189 REVIEW Human ribonuclease P: Subunits, function, and intranuclear localization NAYEF JARROUS Department of Molecular Biology, The Hebrew University–Hadassah Medical School, Jerusalem 91120, Israel ABSTRACT Catalytic complexes of nuclear ribonuclease P (RNase P) ribonucleoproteins are composed of several protein sub- units that appear to have specific roles in enzyme function in tRNA processing. This review describes recent progress made in the characterization of human RNase P, its relationship with the ribosomal RNA processing ribonucleoprotein RNase MRP, and the unexpected evolutionary conservation of its subunits. A new model for the biosynthesis of human RNase P is presented, in which this process is dynamic, transcription-dependent, and implicates functionally distinct nuclear compartments in tRNA biogenesis. Keywords: Cajal bodies; catalytic ribonucleoprotein; nucleolus; RNase MRP; RNase P; tRNA RIBONUCLEOPROTEIN COMPLEXES a ribonucleoprotein complex (Rossmanith et al+, 1995; OF RNase P Rossmanith & Karwan, 1998)+ These opposing find- ings provoke further debate on the biochemical nature Extensive purification analysis of RNase P from HeLa of mitochondrial and other organellar RNase P en- cells has revealed that the nuclear form of this tRNA zymes (Frank & Pace, 1998; Schon, 1999; Altman et al+, -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

Nuclear Domains of the RNA Subunit of Rnase P
Journal of Cell Science 110, 829-837 (1997) 829 Printed in Great Britain © The Company of Biologists Limited 1997 JCS8126 Nuclear domains of the RNA subunit of RNase P Marty R. Jacobson1, Long-Guang Cao1,*, Krishan Taneja2, Robert H. Singer2,†, Yu-li Wang1 and Thoru Pederson1,‡ 1Cell Biology Group, Worcester Foundation for Biomedical Research, Shrewsbury, MA 01545, USA 2Department of Cell Biology, University of Massachusetts Medical Center, Worcester, MA 01655, USA *Present address: Department of Molecular Genetics and Microbiology, University of Florida, PO Box 100266, Gainesville, FL 32610, USA †Present address: Department of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, NY 10461-1975, USA ‡Author for correspondence (e-mail: [email protected]) SUMMARY The ribonucleoprotein enzyme RNase P catalyzes the 5′ microinjection. In contrast, a truncated RNase P RNA con- processing of pre-transfer RNA, and has also recently been taining the To binding domain but lacking nucleotides 89- implicated in pre-ribosomal RNA processing. In the 341 became rapidly localized in nucleoli following nuclear present investigation, in situ hybridization revealed that microinjection. However, unlike the full-length RNase P RNase P RNA is present throughout the nucleus of RNA, this 3′ truncated RNA remained stably associated mammalian cells. However, rhodamine-labeled human with the nucleoli and did not translocate to the nucleo- RNase P RNA microinjected into the nucleus of rat kidney plasm. These results suggest a nucleolar phase in the mat- (NRK) epithelial cells or human (HeLa) cells initially uration, ribonucleoprotein assembly or function of RNase localized in nucleoli, and subsequently became more evenly P RNA, mediated at least in part by the nucleolar To distributed throughout the nucleus, similar to the steady- antigen.