Signature Redacted Certified By: Ronald T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
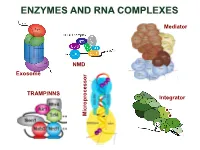
Enzymes and Rna Complexes
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

Ribonuclease A
Chem. Rev. 1998, 98, 1045−1065 1045 Ribonuclease A Ronald T. Raines Departments of Biochemistry and Chemistry, University of WisconsinsMadison, Madison, Wisconsin 53706 Received October 10, 1997 (Revised Manuscript Received January 12, 1998) Contents I. Introduction 1045 II. Heterologous Production 1046 III. Structure 1046 IV. Folding and Stability 1047 A. Disulfide Bond Formation 1047 B. Prolyl Peptide Bond Isomerization 1048 V. RNA Binding 1048 A. Subsites 1048 B. Substrate Specificity 1049 C. One-Dimensional Diffusion 1049 D. Processive Catalysis 1050 VI. Substrates 1050 VII. Inhibitors 1051 Ronald T. Raines was born in 1958 in Montclair, NJ. He received Sc.B. VIII. Reaction Mechanism 1052 degrees in chemistry and biology from the Massachusetts Institute of A. His12 and His119 1053 Technology. At M.I.T., he worked with Christopher T. Walsh to reveal the reaction mechanisms of pyridoxal 5′-phosphate-dependent enzymes. B. Lys41 1054 Raines was a National Institutes of Health predoctoral fellow in the C. Asp121 1055 chemistry department at Harvard University. There, he worked with D. Gln11 1056 Jeremy R. Knowles to elucidate the reaction energetics of triosephosphate IX. Reaction Energetics 1056 isomerase. Raines was a Helen Hay Whitney postdoctoral fellow in the biochemistry and biophysics department at the University of California, A. Transphosphorylation versus Hydrolysis 1056 San Francisco. At U.C.S.F., he worked with William J. Rutter to clone, B. Rate Enhancement 1057 express, and mutate the cDNA that codes for ribonuclease A. Raines X. Ribonuclease S 1058 then joined the faculty of the biochemistry department at the University s A. S-Protein−S-Peptide Interaction 1058 of Wisconin Madison, where he is now associate professor of biochem- istry and chemistry. -

Characterization of the Mammalian RNA Exonuclease 5/NEF-Sp As a Testis-Specific Nuclear 3′′′′′ → 5′′′′′ Exoribonuclease
Downloaded from rnajournal.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Characterization of the mammalian RNA exonuclease 5/NEF-sp as a testis-specific nuclear 3′′′′′ → 5′′′′′ exoribonuclease SARA SILVA,1,2 DAVID HOMOLKA,1 and RAMESH S. PILLAI1 1Department of Molecular Biology, University of Geneva, CH-1211 Geneva 4, Switzerland 2European Molecular Biology Laboratory, Grenoble Outstation, 38042, France ABSTRACT Ribonucleases catalyze maturation of functional RNAs or mediate degradation of cellular transcripts, activities that are critical for gene expression control. Here we identify a previously uncharacterized mammalian nuclease family member NEF-sp (RNA exonuclease 5 [REXO5] or LOC81691) as a testis-specific factor. Recombinant human NEF-sp demonstrates a divalent metal ion-dependent 3′′′′′ → 5′′′′′ exoribonuclease activity. This activity is specific to single-stranded RNA substrates and is independent of their length. The presence of a 2′′′′′-O-methyl modification at the 3′′′′′ end of the RNA substrate is inhibitory. Ectopically expressed NEF-sp localizes to the nucleolar/nuclear compartment in mammalian cell cultures and this is dependent on an amino-terminal nuclear localization signal. Finally, mice lacking NEF-sp are viable and display normal fertility, likely indicating overlapping functions with other nucleases. Taken together, our study provides the first biochemical and genetic exploration of the role of the NEF-sp exoribonuclease in the mammalian genome. Keywords: NEF-sp; LOC81691; Q96IC2; REXON; RNA exonuclease 5; REXO5; 2610020H08Rik INTRODUCTION clease-mediated processing to create their final 3′ ends: poly(A) tails of most mRNAs or the hairpin structure of Spermatogenesis is the process by which sperm cells are replication-dependent histone mRNAs (Colgan and Manley produced in the male germline. -

Protein Engineering to Exploit and Explore Bovine Secretory Ribonucleases
PROTEIN ENGINEERING TO EXPLOIT A N D EXPLORE BOVINE SECRETORY RIBONUCLEASES by JIN-SOO KIM A thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biochemistry) at the UNIVERSITY OF WISCONSIN-MADISON 1994 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGEMENTS I would like to thank Dr. Ronald T. Raines for his advice and support. His scientific insight has been very helpful throughout this work. I would also like to thank the entire Raines group for their friendship and companionship. I am grateful to Dr. J. Soucek and Dr. J. Matousek for their collaboration with us, which has been a valuable part of the BS-RNase research. I thank Dr. M. Karpeisky for suggesting the protein fusion project, and Dr. G. D'Alessio and Dr. L. Mazzarella for providing the coordinates of BS-RNase. I have been generously supported by Steenbock predoctoral fellowship from the Department of Biochemistry. Finally, I thank my parents, who have encouraged (or at least not discouraged) me to pursue a career in science since I was a kid. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission ABSTRACT PROTEIN ENGINEERING TO EXPLOIT AND EXPLORE BOVINE SECRETORY RIBONUCLEASES Jin-Soo Kim Under the supervision of Dr. Ronald T. Raines at the University of Wisconsin-Madison Ribonuclease S-peptide (residues 1-20) and S-protein (residues 21- 124) are the enzymatically inactive products of the limited digestion of bovine pancreatic ribonuclease A (RNase A) by subtilisin. S-Peptide binds S-protein with high affinity to form RNase S, which has full enzymatic activity. -

Insights Into the Structure and Architecture of the CCR4–NOT Complex
REVIEW ARTICLE published: 16 May 2014 doi: 10.3389/fgene.2014.00137 Insights into the structure and architecture of the CCR4–NOT complex Kun Xu 1,2 ,Yuwei Bai 1, Aili Zhang 1,2, Qionglin Zhang 2 and Mark G. Bartlam1,2* 1 State Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin, China 2 College of Life Sciences, Nankai University, Tianjin, China Edited by: The CCR4–NOT complex is a highly conserved, multifunctional machinery with a general Martine Anne Collart, University of role in controlling mRNA metabolism. It has been implicated in a number of different Geneva, Switzerland aspects of mRNA and protein expression, including mRNA degradation, transcription Reviewed by: initiation and elongation, ubiquitination, and protein modification. The core CCR4–NOT Walter Lukiw, Louisiana State University, USA complex is evolutionarily conserved and consists of at least three NOT proteins and two Sebastiaan Winkler, University of catalytic subunits.The L-shapedcomplex is characterized by two functional modules bound Nottingham, UK to the CNOT1/Not1 scaffold protein: the deadenylase or nuclease module containing *Correspondence: two enzymes required for deadenylation, and the NOT module. In this review, we will Mark G. Bartlam, College of Life summarize the currently available information regarding the three-dimensional structure Sciences, Nankai University, 94 Weijin Road, Tianjin 300071, China and assembly of the CCR4–NOT complex, in order to provide insight into its roles in mRNA e-mail: [email protected] degradation and other biological processes. Keywords: CCR4–NOT complex, mRNA decay, poly(A), deadenylation, NOT module, protein structure INTRODUCTION 0.9–1.2 MDa in size (Liu et al., 1998). -

A Quantitative Proteomics Investigation of Cold Adaptation in the Marine Bacterium, Sphingopyxis Alaskensis
A quantitative proteomics investigation of cold adaptation in the marine bacterium, Sphingopyxis alaskensis Thesis submitted in partial fulfilment of the requirements for the Degree of Doctor of Philosophy (Ph.D.) Lily L. J. Ting School of Biotechnology and Biomolecular Sciences University of New South Wales January 2010 COPYRIGHT STATEMENT ‘I hereby grant the University of New South Wales or its agents the right to archive and to make available my thesis or dissertation in whole or part in the University libraries in all forms of media, now or here after known, subject to the provisions of the Copyright Act 1968. I retain all proprietary rights, such as patent rights. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation. I also authorise University Microfilms to use the 350 word abstract of my thesis in Dissertation Abstract International (this is applicable to doctoral theses only). I have either used no substantial portions of copyright material in my thesis or I have obtained permission to use copyright material; where permission has not been granted I have applied/will apply for a partial restriction of the digital copy of my thesis or dissertation.' Signed ……………………………………………........................... 21st April, 2010 Date ……………………………………………........................... AUTHENTICITY STATEMENT ‘I certify that the Library deposit digital copy is a direct equivalent of the final officially approved version of my thesis. No emendation of content has occurred and if there are any minor variations in formatting, they are the result of the conversion to digital format.’ Signed ……………………………………………........................... 21st April, 2010 Date …………………………………………….......................... -

The Rnase III Family: a Conserved Structure and Expanding Functions in Eukaryotic Dsrna Metabolism
Curr. Issues Mol. Biol. (2001) 3(4): 71-78. The Eukaryotic RNase III 71 The RNase III Family: A Conserved Structure and Expanding Functions in Eukaryotic dsRNA Metabolism Bruno Lamontagne, Stéphanie Larose, Jim Boulanger, family (Figure 1). In bacteria, RNase III exists in one form and Sherif Abou Elela* characterized by a classical RNA binding domain and a nuclease domain (Nicholson, 1999). In contrast, eukaryotic Département de Microbiologie et d’Infectiologie, Faculté RNase III exists in three isoforms that share the basic de Médecine, Université de Sherbrooke, Sherbrooke, dsRBD but differ in the number of nuclease domains and Québec, Canada J1H 5N4 in the composition of the N-terminal domain (Filippov et al., 2000; Jacobsen et al., 1999; Lamontagne et al., 2000). The first form contains three domains; the dsRBD, the Abstract nuclease domain, and an additional uniquely eukaryotic N-terminal domain required for correct protein conformation The last few years have witnessed the appreciation of and efficient RNA cleavage (Lamontagne et al., 2000). The dsRNA as a regulator of gene expression, a potential second form exhibits in addition to these three domains a antiviral agent, and a tumor suppressor. However, in second nuclease motif at the protein N-terminus (Wu et spite of these clear effects on the cell function, the al., 2000). Finally, the third form of eukaryotic RNase III mechanism that controls dsRNA maturation and contains in addition to the three main eukaryotic domains stability remains unknown. Recently, the discovery of a fourth distinct helicase domain (Jacobsen et al., 1999; eukaryotic orthologues of the bacterial dsRNA specific Rotondo and Frendewey, 1996). -

Drosophila Melanogaster Dis3 Is a Dynamic Endo- and 3’Æ5’
DROSOPHILA MELANOGASTER DIS3 IS A DYNAMIC ENDO- AND 3’Æ5’ EXORIBONUCLEASE By MEGAN CHRISTINE MAMOLEN Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Dissertation advisor: Erik D. Andrulis, Ph.D. Department of Molecular Biology and Microbiology CASE WESTERN RESERVE UNIVERSITY August, 2010 We hereby approve the thesis/dissertation of ___________________Megan Mamolen_____________________ candidate for the ____________Ph.D.________________degree *. (signed)_________________Dr. Jonathan Karn_________________ (chair of the committee) _____________________Dr. Peter Harte______________________ ___________________Dr. Alan Tartakoff_____________________ _____________________Dr. Erik Andrulis_____________________ (date) _______6-15-10________________ *We also certify that written approval has been obtained for any proprietary material contained therein. 2 Copyright © 2010 by Megan Christine Mamolen All rights reserved 3 This work is dedicated to my husband and best friend, Mike Smolko. Thank you for encouraging me to believe in myself. This is only the beginning of a wonderful journey together. 4 Table of Contents Table of Contents ................................................................................................................ 5 List of Tables .................................................................................................................... 10 List of Figures .................................................................................................................. -

Structural Characterization of Proteinaceous Rnase P from Arabidopsis Thaliana Franziska Pinker
Structural characterization of proteinaceous RNase P from Arabidopsis thaliana Franziska Pinker To cite this version: Franziska Pinker. Structural characterization of proteinaceous RNase P from Arabidopsis thaliana. Biologie végétale. Université de Strasbourg, 2014. Français. NNT : 2014STRAJ093. tel-01236246 HAL Id: tel-01236246 https://tel.archives-ouvertes.fr/tel-01236246 Submitted on 1 Dec 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITÉ DE STRASBOURG ÉCOLE DOCTORALE des Sciences de la Vie et de la Santé Institut de Biologie Moléculaire des Plantes - IBMP THÈSE présentée par : Franziska PINKER soutenue le : 15 septembre 2014 pour obtenir le grade de : !"#$%&'($')R%+ iversité de Strasbourg Discipline/ Spécialité : Sciences du Vivant /Biochimie, Biologie Moléculaire et Structurale Structural characterization of proteinaceous RNase P from Arabidopsis thaliana THÈSE dirigée par : M. Claude SAUTER Dr, Université de Strasbourg M. Philippe GIEGE Dr, Université de Strasbourg RAPPORTEURS : M. Walter ROSSMANITH Dr, Université de Vienne Mme Emmanuelle SCHMITT Dr, Ecole Polytechnique AUTRES MEMBRES DU JURY : Mme Anita MARCHFELDER Professeur, !"#$%&"' )*R ,- Mme Pascale ROMBY Dr, Université de Strasbourg Fur¨ meine Familie Acknowledgements First of all I would like to thank the members of my jury, Dr. -
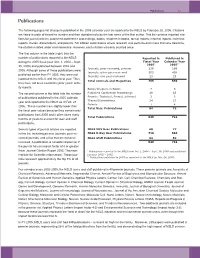
NSLS Users and Staff Published in 40 Premier Journals
Publications 7-3 Publications The following pages list all papers published in the 2005 calendar year as reported to the NSLS by February 28, 2006. Citations are listed in order of beamline number and then alphabetically by the last name of the first author. This list contains reported cita- tions for journal articles, published conference proceedings, books, chapters in books, formal reports, informal reports, technical reports, theses, dissertations, and patents. For citation submissions where research was performed on more than one beamline, the citation is listed under each beamline. However, each citation was only counted once. The first column in the table (right) lists the number of publications reported to the NSLS Reported in Published in during the 2005 fiscal year (Oct. 1, 2004 – Sept. Fiscal Year Calendar Year 2005* 2005** 30, 2005) and published between 2002 and Journals, peer-reviewed, premier 239 189 2005. Although some of these publications were Journals, other peer-reviewed 503 438 published earlier than FY 2005, they were not Journals, non peer-reviewed 23 23 reported to the NSLS until this fiscal year. Thus, Total Journals and Magazines 765 650 they have not been counted in prior years’ activ- ity reports. Books/Chapters in Books 7 5 The second column in the table lists the number Published Conference Proceedings 49 43 of publications published in the 2005 calendar Reports: Technical, Formal, Informal 3 3 Theses/Dissertations 24 17 year and reported to the NSLS as of Feb. 28, Patents 1 4 2006. These numbers are slightly lower than Total Misc. Publications 84 72 the fiscal year values because they contain only publications from 2005 and it often takes many Total Publications 849 722 months or years to account for user and staff publications. -

Regulators of HIV-1 Reverse Transcription
Viruses 2009, 1, 873-894; doi:10.3390/v1030873 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Reverse Transcriptase and Cellular Factors: Regulators of HIV-1 Reverse Transcription Kylie Warren 1,2, David Warrilow 1, Luke Meredith 1,3 and David Harrich 1,3,* 1 Division of Infectious Diseases, Queensland Institute of Medical Research, Brisbane, QLD, Australia; E-Mails: [email protected] (K.W.); [email protected] (D.W.); [email protected] (L.M.) 2 School of Natural Sciences, University of Western Sydney, Hawkesbury, NSW, Australia 3 Griffith Medical Research College, a joint program of Griffith University and the Queensland Institute of Medical Research, QIMR, Herston, QLD, 4006, Australia * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +61-7-3845-36791; Fax: +61-7-3362-0107. Received: 30 September 2009; in revised form: 6 November 2009 / Accepted: 9 November 2009 / Published: 10 November 2009 Abstract: There is ample evidence that synthesis of HIV-1 proviral DNA from the viral RNA genome during reverse transcription requires host factors. However, only a few cellular proteins have been described in detail that affect reverse transcription and interact with reverse transcriptase (RT). HIV-1 integrase is an RT binding protein and a number of IN-binding proteins including INI1, components of the Sin3a complex, and Gemin2 affect reverse transcription. In addition, recent studies implicate the cellular proteins HuR, AKAP149, and DNA topoisomerase I in reverse transcription through an interaction with RT. In this review we will consider interactions of reverse transcription complex with viral and cellular factors and how they affect the reverse transcription process. -

Trna Biology Charges to the Front
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW tRNA biology charges to the front Eric M. Phizicky1,4 and Anita K. Hopper2,3 1Department of Biochemistry and Biophysics, Center for RNA Biology, University of Rochester School of Medicine, Rochester, New York 14642, USA; 2Department of Molecular Genetics, Center for RNA Biology, Ohio State University, Columbus, Ohio 43210, USA tRNA biology has come of age, revealing an unprecedented Multiple layers of regulation of tRNA transcription level of understanding and many unexpected discoveries tRNA and rRNA genes are highly transcribed, leading to along the way. This review highlights new findings on the the production in yeast of ;3 million tRNAs per gener- diverse pathways of tRNA maturation, and on the forma- ation and 300,000 ribosomes (Waldron and Lacroute tion and function of a number of modifications. Topics of 1975), compared with about 60,000 mRNAs (Ares et al. special focus include the regulation of tRNA biosynthesis, 1999). Because of the energy devoted to tRNA and rRNA quality control tRNA turnover mechanisms, widespread transcription, and because of the required coordination of tRNA cleavage pathways activated in response to stress tRNA and ribosome function, tRNA transcription via and other growth conditions, emerging evidence of signal- RNA polymerase III (Pol III) and rRNA transcription via ing pathways involving tRNA and cleavage fragments, and Pol I need to be coordinated and regulated in response to the sophisticated intracellular tRNA trafficking that oc- cellular nutrient availability and other environmental curs during and after biosynthesis. information. The consequences of inappropriate regula- tion of tRNA transcription have been underscored by the tRNA biogenesis involves the synthesis of the initial results of Marshall et al.