A Quantitative Proteomics Investigation of Cold Adaptation in the Marine Bacterium, Sphingopyxis Alaskensis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Aerobic Anoxygenic Phototrophic Bacterium Sphingomonas Sp
microorganisms Article Characterization of the Aerobic Anoxygenic Phototrophic Bacterium Sphingomonas sp. AAP5 Karel Kopejtka 1 , Yonghui Zeng 1,2, David Kaftan 1,3 , Vadim Selyanin 1, Zdenko Gardian 3,4 , Jürgen Tomasch 5,† , Ruben Sommaruga 6 and Michal Koblížek 1,* 1 Centre Algatech, Institute of Microbiology, Czech Academy of Sciences, 379 81 Tˇreboˇn,Czech Republic; [email protected] (K.K.); [email protected] (Y.Z.); [email protected] (D.K.); [email protected] (V.S.) 2 Department of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, 1871 Frederiksberg C, Denmark 3 Faculty of Science, University of South Bohemia, 370 05 Ceskˇ é Budˇejovice,Czech Republic; [email protected] 4 Institute of Parasitology, Biology Centre, Czech Academy of Sciences, 370 05 Ceskˇ é Budˇejovice,Czech Republic 5 Research Group Microbial Communication, Technical University of Braunschweig, 38106 Braunschweig, Germany; [email protected] 6 Laboratory of Aquatic Photobiology and Plankton Ecology, Department of Ecology, University of Innsbruck, 6020 Innsbruck, Austria; [email protected] * Correspondence: [email protected] † Present Address: Department of Molecular Bacteriology, Helmholtz-Centre for Infection Research, 38106 Braunschweig, Germany. Abstract: An aerobic, yellow-pigmented, bacteriochlorophyll a-producing strain, designated AAP5 Citation: Kopejtka, K.; Zeng, Y.; (=DSM 111157=CCUG 74776), was isolated from the alpine lake Gossenköllesee located in the Ty- Kaftan, D.; Selyanin, V.; Gardian, Z.; rolean Alps, Austria. Here, we report its description and polyphasic characterization. Phylogenetic Tomasch, J.; Sommaruga, R.; Koblížek, analysis of the 16S rRNA gene showed that strain AAP5 belongs to the bacterial genus Sphingomonas M. Characterization of the Aerobic and has the highest pairwise 16S rRNA gene sequence similarity with Sphingomonas glacialis (98.3%), Anoxygenic Phototrophic Bacterium Sphingomonas psychrolutea (96.8%), and Sphingomonas melonis (96.5%). -

Acetyl-Coa Synthetase 3 Promotes Bladder Cancer Cell Growth Under Metabolic Stress Jianhao Zhang1, Hongjian Duan1, Zhipeng Feng1,Xinweihan1 and Chaohui Gu2
Zhang et al. Oncogenesis (2020) 9:46 https://doi.org/10.1038/s41389-020-0230-3 Oncogenesis ARTICLE Open Access Acetyl-CoA synthetase 3 promotes bladder cancer cell growth under metabolic stress Jianhao Zhang1, Hongjian Duan1, Zhipeng Feng1,XinweiHan1 and Chaohui Gu2 Abstract Cancer cells adapt to nutrient-deprived tumor microenvironment during progression via regulating the level and function of metabolic enzymes. Acetyl-coenzyme A (AcCoA) is a key metabolic intermediate that is crucial for cancer cell metabolism, especially under metabolic stress. It is of special significance to decipher the role acetyl-CoA synthetase short chain family (ACSS) in cancer cells confronting metabolic stress. Here we analyzed the generation of lipogenic AcCoA in bladder cancer cells under metabolic stress and found that in bladder urothelial carcinoma (BLCA) cells, the proportion of lipogenic AcCoA generated from glucose were largely reduced under metabolic stress. Our results revealed that ACSS3 was responsible for lipogenic AcCoA synthesis in BLCA cells under metabolic stress. Interestingly, we found that ACSS3 was required for acetate utilization and histone acetylation. Moreover, our data illustrated that ACSS3 promoted BLCA cell growth. In addition, through analyzing clinical samples, we found that both mRNA and protein levels of ACSS3 were dramatically upregulated in BLCA samples in comparison with adjacent controls and BLCA patients with lower ACSS3 expression were entitled with longer overall survival. Our data revealed an oncogenic role of ACSS3 via regulating AcCoA generation in BLCA and provided a promising target in metabolic pathway for BLCA treatment. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction acetyl-CoA synthetase short chain family (ACSS), which In cancer cells, considerable number of metabolic ligates acetate and CoA6. -

3-Iodo-Alpha-Methyl-L- Tyrosine
A Strategyfor the Study of CerebralAmino Acid Transport Using Iodine-123-Labeled Amino Acid Radiopharmaceutical: 3-Iodo-alpha-methyl-L- tyrosine Keiichi Kawai, Yasuhisa Fujibayashi, Hideo Saji, Yoshiharu Yonekura, Junji Konishi, Akiko Kubodera, and Akira Yokoyama Faculty ofPharmaceutical Sciences, Science Universityof Tokyo, Tokyo, Japan and School ofMedicine and Faculty of Pharmaceutical Sciences, Kyolo University, Kyoto, Japan In the present work, a search for radioiodinated tyrosine We examined the brain accumulation of iodine-i 23-iodo-al derivatives for the cerebral tyrosine transport is attempted. pha-methyl-L-tyrosine (123I-L-AMT)in mice and rats. l-L-AMT In the screening process, in vitro accumulation studies in showed high brain accumulation in mice, and in rats; rat brain rat brain slices, measurement ofbrain uptake index (BUI), uptake index exceeded that of 14C-L-tyrosine.The brain up and in vivo mouse biodistribution are followed by the take index and the brain slice studies indicated the affinity of analysis of metabolites. In a preliminary study, the radio l-L-AMT for earner-mediatedand stereoselective active trans iodinated monoiodotyrosine in its L- and D-form (L-MIT port systems, respectively; both operating across the blood and D-MIT) and its noniodinated counterpart (‘4C-L- brain barrier and cell membranes of the brain. The tissue tyrosine) are tested. homogenate analysis revealed that most of the accumulated radioactivity belonged to intact l-L-AMT, an indication of its The initial part of the work provided the basis for a stability. Thus, 1231-L-AMTappears to be a useful radiophar radioiodinated tyrosine derivative to be used for the meas maceutical for the selective measurement of cerebral amino urement of cerebral tyrosine transport. -

Cold Shock Response in Mammalian Cells
J. Mol. Microbiol. Biotechnol. (1999) 1(2): 243-255. Cold Shock ResponseJMMB in Mammalian Symposium Cells 243 Cold Shock Response in Mammalian Cells Jun Fujita* Less is known about the cold shock responses. In microorganisms, cold stress induces the synthesis of Department of Clinical Molecular Biology, several cold-shock proteins (Jones and Inouye, 1994). A Faculty of Medicine, Kyoto University, Kyoto, Japan variety of plant genes are known to be induced by cold stress, and are thought to be involved in the stress tolerance of the plant (Shinozaki and Yamaguchi-Shinozaki, 1996; Abstract Hughes et al., 1999). The response to cold stress in mammals, however, has attracted little attention except in Compared to bacteria and plants, the cold shock a few areas such as adaptive thermogenesis, cold response has attracted little attention in mammals tolerance, and storage of cells and organs. Recently, except in some areas such as adaptive thermogenesis, hypothermia is gaining popularity in emergency clinics as cold tolerance, storage of cells and organs, and a novel therapeutic modality for brain damages. In addition, recently, treatment of brain damage and protein low temperature cultivation has been dicussed as a method production. At the cellular level, some responses of to improve heterologous protein production in mammalian mammalian cells are similar to microorganisms; cold cells (Giard et al., 1982). stress changes the lipid composition of cellular Adaptive thermogenesis refers to a component of membranes, and suppresses the rate of protein energy expenditure, which is separable from physical synthesis and cell proliferation. Although previous activity. It can be elevated in response to changing studies have mostly dealt with temperatures below environmental conditions, most notably cold exposure and 20°C, mild hypothermia (32°C) can change the cell’s overfeeding. -

ADDENDUM SHEET America's Boating Course
ADDENDUM SHEET SM America’s Boating Course - 2001 Edition Homeland Security Measures Boaters must be aware of rules and guidelines regarding homeland security measures. The following are steps that boaters should take to protect our country and are a direct result of the terrorist attacks of 11 September 2001. Keep your distance from all military vessels, cruise lines, or commercial shipping: • All vessels must proceed at a no-wake speed when within a Protection Zone (which extends 500 yards around U.S. naval vessels). • Non-military vessels are not allowed to enter within 100 yards of a U.S. naval vessel, whether underway or moored, unless authorized by an official patrol. The patrol may be either Coast Guard or Navy. • Violating the Naval Vessel Protection Zone is a felony offense, punishable by up to six years imprisonment and / or up to $250,000 in fines. Observe and avoid all security zones. Avoid commercial port operation areas. Avoid restricted areas near: • Dams • Naval ship yards • Power plants • Dry docks Do not stop or anchor beneath bridges or in channels. Keep your boat locked when not using it, including while at temporary docks, such as yacht clubs, restaurants, marinas, shopping, etc. When storing your boat disable the engine. If on a trailer, immobilize it so it cannot be moved. Keep a sharp eye out for anything that looks peculiar or out of the ordinary, and report it to the Coast Guard, port or marine security. When boating within a foreign country make certain that you check-in with the foreign country’s Customs Service upon entering the country and with the USA Customs Service and/or Immigration and Naturalization Service upon returning. -

(12) United States Patent (10) Patent No.: US 8,603,824 B2 Ramseier Et Al
USOO8603824B2 (12) United States Patent (10) Patent No.: US 8,603,824 B2 Ramseier et al. (45) Date of Patent: Dec. 10, 2013 (54) PROCESS FOR IMPROVED PROTEIN 5,399,684 A 3, 1995 Davie et al. EXPRESSION BY STRAIN ENGINEERING 5,418, 155 A 5/1995 Cormier et al. 5,441,934 A 8/1995 Krapcho et al. (75) Inventors: Thomas M. Ramseier, Poway, CA 5,508,192 A * 4/1996 Georgiou et al. .......... 435/252.3 (US); Hongfan Jin, San Diego, CA 5,527,883 A 6/1996 Thompson et al. (US); Charles H. Squires, Poway, CA 5,558,862 A 9, 1996 Corbinet al. 5,559,015 A 9/1996 Capage et al. (US) 5,571,694 A 11/1996 Makoff et al. (73) Assignee: Pfenex, Inc., San Diego, CA (US) 5,595,898 A 1/1997 Robinson et al. 5,610,044 A 3, 1997 Lam et al. (*) Notice: Subject to any disclaimer, the term of this 5,621,074 A 4/1997 Bjorn et al. patent is extended or adjusted under 35 5,622,846 A 4/1997 Kiener et al. 5,641,671 A 6/1997 Bos et al. U.S.C. 154(b) by 471 days. 5,641,870 A 6/1997 Rinderknecht et al. 5,643,774 A 7/1997 Ligon et al. (21) Appl. No.: 11/189,375 5,662,898 A 9/1997 Ligon et al. (22) Filed: Jul. 26, 2005 5,677,127 A 10/1997 Hogan et al. 5,683,888 A 1 1/1997 Campbell (65) Prior Publication Data 5,686,282 A 11/1997 Lam et al. -

Applications of Novosphingobium Puteolanum Pp1y
A STUDY OF THE BIOTECHNOLOGICAL APPLICATIONS OF NOVOSPHINGOBIUM PUTEOLANUM PP1Y. Dr. Luca Troncone Dottorato in Scienze Biotecnologiche – XXIV° ciclo Indirizzo Biotecnologie Industriali e Molecolari Università di Napoli Federico II Dottorato in Scienze Biotecnologiche – XXIV° ciclo Indirizzo Biotecnologie Industriali e Molecolari Università di Napoli Federico II A STUDY OF THE BIOTECHNOLOGICAL APPLICATIONS OF NOVOSPHINGOBIUM PUTEOLANUM PP1Y. Dr. Luca Troncone Dottorando: Dr. Luca Troncone Relatore: Prof. Alberto Di Donato Coordinatore: Prof. Giovanni Sannia A zia Nanna Index INDEX RIASSUNTO pag. 3 SUMMARY pag. 8 I. INTRODUCTION pag. 9 1.1. Antropic pollution and bioremediation. 1.2. Microbial biofilm. 1.3. Bioremediation and biofilm. 1.4. Novosphingobium puteolanum PP1Y. 1.5. Aim of the project. II. MATERIALS & METHODS pag. 23 2.1. Culture Media. 2.2. PAH-Agar Plates. 2.3. Optimal Salt Concentration, pH and Temperature for Growth of Strain PP1Y. 2.4. Growth on Fuels. 2.5. Growth on Single Hydrocarbons. 2.6. Phase Contrast Microscopy. 2.7. Removal of Oil-Dissolved Aromatic Hydrocarbons by Strain PP1Y. 2.8. Removal of Aromatic Hydrocarbons from polluted soils: 2.8.1. Growing conditions; 2.8.2. Preparation of microcosms; 2.8.3. Removal of aromatic hydrocarbons from soil by strain PP1Y. 2.9. Heavy metals resistance. 2.10. Analysis of the Extracellular Products: 2.10.1. Proteins analysis: 2.10.1.1. Mass spectrometric analysis. 2.10.2. Carbohydrate analysis: 2.10.2.1. Acetylated methyl glycosides. 2.10.3. Emulsification procedures. 2.11. Genome Analysis. 1 Index 2.12. Other Methods. III. RESULTS & DISCUSSION pag. 31 3.1. Characterization of Novosphingobium puteolanum PP1Y. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -
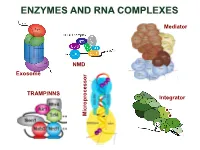
Enzymes and Rna Complexes
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

Autonomic Conflict: a Different Way to Die During Cold Water Immersion
J Physiol 590.14 (2012) pp 3219–3230 3219 TOPICAL REVIEW ‘Autonomic conflict’: a different way to die during cold water immersion? Michael J. Shattock1 and Michael J. Tipton2 1Cardiovascular Division, King’s College London, London, UK 2Extreme Environments Laboratory, Department of Sports and Exercise Science, University of Portsmouth, Portsmouth, UK Abstract Cold water submersion can induce a high incidence of cardiac arrhythmias in healthy volunteers. Submersion and the release of breath holding can activate two powerful and antagonistic responses: the ‘cold shock response’ and the ‘diving response’.The former involves the activation of a sympathetically driven tachycardia while the latter promotes a parasympathetically mediated bradycardia. We propose that the strong and simultaneous activation of the two limbs of the autonomic nervous system (‘autonomic conflict’) may account for these arrhythmias and may, in some vulnerable individuals, be responsible for deaths that have previously wrongly been ascribed to drowning or hypothermia. In this review, we consider the evidence supporting this claim and also hypothesise that other environmental triggers may induce autonomic conflict and this may be more widely responsible for sudden death in individuals with other predisposing conditions. (Received 6 February 2012; accepted after revision 27 April 2012; first published online 30 April 2012) Corresponding author M. Shattock: Cardiovascular Division, King’s College London, The Rayne Institute, Lambeth Wing, St Thomas’ Hospital, London SE1 7EH, UK. Email: [email protected] Introduction: do all drowning victims drown? on average, we lose about one child a week. Historically, death in cold water was generally ascribed to hypothermia; In most countries of the world, immersion represents the more recently, description of the initial ‘cold shock’ second most common cause of accidental death in children response (Tipton, 1989b) to immersion and other factors and the third in adults (Bierens et al. -

Nucleotides and Nucleic Acids
Nucleotides and Nucleic Acids Energy Currency in Metabolic Transactions Essential Chemical Links in Response of Cells to Hormones and Extracellular Stimuli Nucleotides Structural Component Some Enzyme Cofactors and Metabolic Intermediate Constituents of Nucleic Acids: DNA & RNA Basics about Nucleotides 1. Term Gene: A segment of a DNA molecule that contains the information required for the synthesis of a functional biological product, whether protein or RNA, is referred to as a gene. Nucleotides: Nucleotides have three characteristic components: (1) a nitrogenous (nitrogen-containing) base, (2) a pentose, and (3) a phosphate. The molecule without the phosphate groups is called a nucleoside. Oligonucleotide: A short nucleic acid is referred to as an oligonucleotide, usually contains 50 or fewer nucleotides. Polynucleotide: Polymers containing more than 50 nucleotides is usually referred to as polynucleotide. General structure of nucleotide, including a phosphate group, a pentose and a base unit (either Purine or Pyrimidine). Major purine and Pyrimidine bases of nucleic acid The roles of RNA and DNA DNA: a) Biological Information Storage, b) Biological Information Transmission RNA: a) Structural components of ribosomes and carry out the synthesis of proteins (Ribosomal RNAs: rRNA); b) Intermediaries, carry genetic information from gene to ribosomes (Messenger RNAs: mRNA); c) Adapter molecules that translate the information in mRNA to proteins (Transfer RNAs: tRNA); and a variety of RNAs with other special functions. 1 Both DNA and RNA contain two major purine bases, adenine (A) and guanine (G), and two major pyrimidines. In both DNA and RNA, one of the Pyrimidine is cytosine (C), but the second major pyrimidine is thymine (T) in DNA and uracil (U) in RNA. -

Changes in the Sclerotinia Sclerotiorum Transcriptome During Infection of Brassica Napus
Seifbarghi et al. BMC Genomics (2017) 18:266 DOI 10.1186/s12864-017-3642-5 RESEARCHARTICLE Open Access Changes in the Sclerotinia sclerotiorum transcriptome during infection of Brassica napus Shirin Seifbarghi1,2, M. Hossein Borhan1, Yangdou Wei2, Cathy Coutu1, Stephen J. Robinson1 and Dwayne D. Hegedus1,3* Abstract Background: Sclerotinia sclerotiorum causes stem rot in Brassica napus, which leads to lodging and severe yield losses. Although recent studies have explored significant progress in the characterization of individual S. sclerotiorum pathogenicity factors, a gap exists in profiling gene expression throughout the course of S. sclerotiorum infection on a host plant. In this study, RNA-Seq analysis was performed with focus on the events occurring through the early (1 h) to the middle (48 h) stages of infection. Results: Transcript analysis revealed the temporal pattern and amplitude of the deployment of genes associated with aspects of pathogenicity or virulence during the course of S. sclerotiorum infection on Brassica napus. These genes were categorized into eight functional groups: hydrolytic enzymes, secondary metabolites, detoxification, signaling, development, secreted effectors, oxalic acid and reactive oxygen species production. The induction patterns of nearly all of these genes agreed with their predicted functions. Principal component analysis delineated gene expression patterns that signified transitions between pathogenic phases, namely host penetration, ramification and necrotic stages, and provided evidence for the occurrence of a brief biotrophic phase soon after host penetration. Conclusions: The current observations support the notion that S. sclerotiorum deploys an array of factors and complex strategies to facilitate host colonization and mitigate host defenses. This investigation provides a broad overview of the sequential expression of virulence/pathogenicity-associated genes during infection of B.