Glycerol Dehydrogenase from Gluconobacter Industrius
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

High-Yield Anaerobic Succinate Production by Strategically
Meng et al. Microb Cell Fact (2016) 15:141 DOI 10.1186/s12934-016-0536-1 Microbial Cell Factories RESEARCH Open Access High‑yield anaerobic succinate production by strategically regulating multiple metabolic pathways based on stoichiometric maximum in Escherichia coli Jiao Meng1,2,3†, Baiyun Wang1,2,3†, Dingyu Liu1,2,3, Tao Chen1,2,3,4, Zhiwen Wang1,2,3* and Xueming Zhao1,2,3 Abstract Background: Succinate has been identified by the U.S. Department of Energy as one of the top 12 building block chemicals, which can be used as a specialty chemical in the agricultural, food, and pharmaceutical industries. Escheri- chia coli are now one of the most important succinate producing candidates. However, the stoichiometric maximum succinate yield under anaerobic conditions through the reductive branch of the TCA cycle is restricted by NADH sup- ply in E. coli. Results: In the present work, we report a rational approach to increase succinate yield by regulating NADH supply via pentose phosphate (PP) pathway and enhancing flux towards succinate. The deregulated genes zwf243 (encod- ing glucose-6-phosphate dehydrogenase) and gnd361 (encoding 6-phosphogluconate dehydrogenase) involved in NADPH generation from Corynebacterium glutamicum were firstly introduced into E. coli for succinate production. Co-expression of beneficial mutated dehydrogenases, which removed feedback inhibition in the oxidative part of the PP pathway, increased succinate yield from 1.01 to 1.16 mol/mol glucose. Three critical genes, pgl (encoding 6-phos- phogluconolactonase), tktA (encoding transketolase) and talB (encoding transaldolase) were then overexpressed to redirect more carbon flux towards PP pathway and further improved succinate yield to 1.21 mol/mol glucose. -

The Diversity of Microbial Aldo/Keto Reductases from Escherichia Coli K12
Lapthorn, A. J., Zhu, X., and Ellis, E. M. (2013) The diversity of microbial aldo/keto reductases from Escherichia coli K12. Chemico-Biological Interactions, 202(1-3), pp. 168-177. (doi:10.1016/j.cbi.2012.10.008) There may be differences between this version and the published version. You are advised to consult the publisher’s version if you wish to cite from it. http://eprints.gla.ac.uk/124204/ Deposited on: 07 October 2016 Enlighten – Research publications by members of the University of Glasgow http://eprints.gla.ac.uk The diversity of microbial aldo/keto reductases from Escherichia coli K12 Adrian J. Lapthorn 1, Xiaofeng Zhu 2,3 and Elizabeth M. Ellis 3 1 School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ 2 College of Life Science and State Key Laboratory of Biotherapy and Cancer Centre, Sichuan University, Chengdu, China 3 Strathclyde Institute of Pharmacy and Biomedical Sciences, 161 Cathedral Street, Glasgow, G4 0RE Corresponding author: Adrian J. Lapthorn School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ Tel: +44 141-330 5940 Fax: +44 141-330 4888 E-mail: [email protected] Key Words: Aldo-Keto reductases, methylglyoxal reductase, 2,5-diketo-D-gluconate reductase, tyrosine auxotrophy suppressor protein, L-glyceraldehyde 3-phosphate reductase, AKR quaternary structure. Abstract The genome of Escherichia coli K12 contains 9 open reading frames encoding aldo/keto reductases (AKR) that are differentially regulated and sequence diverse. A significant amount of data is available for the E. coli AKRs through the availability of gene knockouts and gene expression studies, which adds to the biochemical and kinetic data. -

Engineering of Glycerol Utilization in Gluconobacter Oxydans 621H For
Yan et al. Microb Cell Fact (2018) 17:158 https://doi.org/10.1186/s12934-018-1001-0 Microbial Cell Factories RESEARCH Open Access Engineering of glycerol utilization in Gluconobacter oxydans 621H for biocatalyst preparation in a low‑cost way Jinxin Yan1, Jing Xu1,3, Menghao Cao1, Zhong Li1, Chengpeng Xu1, Xinyu Wang1, Chunyu Yang1, Ping Xu2, Chao Gao1 and Cuiqing Ma1* Abstract Background: Whole cells of Gluconobacter oxydans are widely used in various biocatalytic processes. Sorbitol at high concentrations is commonly used in complex media to prepare biocatalysts. Exploiting an alternative process for preparation of biocatalysts with low cost substrates is of importance for industrial applications. Results: G. oxydans 621H was confrmed to have the ability to grow in mineral salts medium with glycerol, an inevitable waste generated from industry of biofuels, as the sole carbon source. Based on the glycerol utilization mechanism elucidated in this study, the major polyol dehydrogenase (GOX0854) and the membrane-bound alcohol dehydrogenase (GOX1068) can competitively utilize glycerol but play no obvious roles in the biocatalyst prepara- tion. Thus, the genes related to these two enzymes were deleted. Whole cells of G. oxydans ∆GOX1068∆GOX0854 can be prepared from glycerol with a 2.4-fold higher biomass yield than that of G. oxydans 621H. Using whole cells of G. 1 1 oxydans ∆GOX1068∆GOX0854 as the biocatalyst, 61.6 g L− xylonate was produced from 58.4 g L− xylose at a yield of 1 1.05 g g− . Conclusion: This process is an example of efcient preparation of whole cells of G. oxydans with reduced cost. -

Characterization of Glycerol Dehydrogenase from Thermoanaerobacterium Thermosaccharolyticum DSM 571 and GGG Motif Identification
J. Microbiol. Biotechnol. (2016), 26(6), 1077–1086 http://dx.doi.org/10.4014/jmb.1512.12051 Research Article Review jmb Characterization of Glycerol Dehydrogenase from Thermoanaerobacterium thermosaccharolyticum DSM 571 and GGG Motif Identification Liangliang Wang1,2, Jiajun Wang1,2, Hao Shi1,2, Huaxiang Gu1,2, Yu Zhang1,2, Xun Li1,2, and Fei Wang1,2* 1College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, P.R. China 2Jiangsu Key Laboratory of Biomass-Based Green Fuels and Chemicals, Nanjing 210037, P.R. China Received: December 17, 2015 Revised: March 5, 2016 Glycerol dehydrogenases (GlyDHs) are essential for glycerol metabolism in vivo, catalyzing Accepted: March 9, 2016 its reversible reduction to 1,3-dihydroxypropranone (DHA). The gldA gene encoding a putative GlyDH was cloned from Thermoanaerobacterium thermosaccharolyticum DSM 571 (TtGlyDH) and expressed in Escherichia coli. The presence of Mn2+ enhanced its enzymatic 171 254 271 First published online activity by 79.5%. Three highly conserved residues (Asp , His , and His ) in TtGlyDH were March 14, 2016 associated with metal ion binding. Based on an investigation of glycerol oxidation and DHA *Corresponding author reduction, TtGlyDH showed maximum activity towards glycerol at 60°C and pH 8.0 and Phone: +86-25-85427649; towards DHA at 60°C and pH 6.0. DHA reduction was the dominant reaction, with a lower Fax: +86-25-85427649; K of 1.08 ± 0.13 mM and V of 0.0053 ± 0.0001 mM/s, compared with glycerol oxidation, E-mail: [email protected] m(DHA) max with a Km(glycerol) of 30.29 ± 3.42 mM and Vmax of 0.042 ± 0.002 mM/s. -

Molecular Properties of Membrane-Bound FAD-Containing D-Sorbitol Dehydrogenase from Thermotolerant Gluconobacter Frateurii Isolated from Thailand
Biosci. Biotechnol. Biochem., 69 (6), 1120–1129, 2005 Molecular Properties of Membrane-Bound FAD-Containing D-Sorbitol Dehydrogenase from Thermotolerant Gluconobacter frateurii Isolated from Thailand y Hirohide TOYAMA,1; Wichai SOEMPHOL,1 Duangtip MOONMANGMEE,2 Osao ADACHI,1 and Kazunobu MATSUSHITA1 1Department of Biological Chemistry, Faculty of Agriculture, Yamaguchi University, Yamaguchi 753-8515, Japan 2Department of Microbiology, Faculty of Science, King Mongkut’s University of Technology Thonburi, Prachauthit Road., Tungkru, Bangkok 10140, Thailand Received December 24, 2004; Accepted March 14, 2005 There are two types of membrane-bound D-sorbitol ism leads to applications in industry for fermentation of dehydrogenase (SLDH) reported: PQQ–SLDH, having valuable products such as L-sorbose, dihydroxyacetone, pyrroloquinoline quinone (PQQ), and FAD–SLDH, D-gluconate, and keto-D-gluconates.3,4) containing FAD and heme c as the prosthetic groups. L-Sorbose is an important intermediate in the indus- FAD–SLDH was purified and characterized from the trial production of vitamin C. The membrane-bound PQQ–SLDH mutant strain of a thermotolerant Gluco- D-sorbitol dehydrogenase (SLDH) has been considered nobacter frateurii, having molecular mass of 61.5 kDa, to play a main role in L-sorbose fermentation.5) In 52 kDa, and 22 kDa. The enzyme properties were quite Gluconobacter strains, two types of membrane-bound similar to those of the enzyme from mesophilic G. oxy- D-sorbitol dehydrogenases have been purified and well dans IFO 3254. This enzyme was shown to be inducible characterized. One was purified from G. suboxydans var by D-sorbitol, but not PQQ–SLDH. The oxidation IFO 32546) as flavohemoprotein, containing a cova- product of FAD–SLDH from D-sorbitol was identified lently bound FAD having three subunits with molecular as L-sorbose. -

Transcriptional Regulation Mechanisms Involved in Azole Resistance in Candida Species: Focusing on the Transcription Factors Rpn4 and Mrr1
Transcriptional regulation mechanisms involved in azole resistance in Candida species: focusing on the transcription factors Rpn4 and Mrr1 Raquel da Silva Califórnia Thesis to obtain the Master of Science Degree in Biotechnology Supervisor: Prof. Dr. Miguel Nobre Parreira Cacho Teixeira Examination Committee Chairperson: Prof. Dr. Ana Cristina Anjinho Madeira Viegas Supervisor: Prof. Dr. Miguel Nobre Parreira Cacho Teixeira Member of the Committee: Dr. Catarina Isabel Ribeiro Pimentel October 2018 ii Acknowledgements For me the development of this thesis was very challenging and involved a very extensive work, whose purpose would not have been reached without the help of some people I will mention below. First of all, I would like to thank my supervisor Professor Miguel Teixeira for the opportunity given by accepting me in his team and in this project. His tremendous support, guidance and motivation, always available to help, were crucial for the success of this work. I would like to thank Professor Isabel Sá-Correia for giving me the chance to join the Biological Sciences Research Group to develop my master thesis work. The achievement of this thesis required an indispensable help from several parts, which deserve my recognition. For the collaboration in the transcriptomic analysis herein accomplished, I thank Professor Geraldine Butler and her team, from University College of Dublin. For the supply of Candida glabrata mutants used in this work, I have to thank Professor Hiroji Chibana, from University of Chiba, Japan. For the study developed in HPLC analysis of ergosterol levels, I thank also Professor Nuno Mira for his availability and assistance. My gratitude should also be expressed towards my colleague, Pedro Pais, for the great help he has given me throughout this period, always available to help and explain anything. -
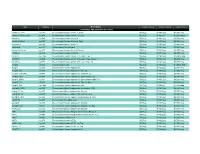
Prospec (Product
Name Catalog # Description Customer Price-A Customer Price-B Customer Price-C CYTOKINES AND GROWTH FACTORS Activin A, Active cyt-145 Recombinant Human Activin-A, Active $50/2µg $130/10µg $3,500/1mg Activin A, Plant-Active cyt-414 Recombinant Human Activin-A Active $50/1µg $130/5µg $1,500/100µg Activin A cyt-569 Recombinant Human Activin-A $50/2µg $130/10µg $2,700/1mg Activin A, Plant cyt-052 Recombinant Human Activin-A, Plant $50/2µg $130/10µg $4,800/1mg mActivin A cyt-146 Recombinant Mouse Activin-A $50/2µg $130/10µg $3,500/1mg rActivin A cyt-147 Recombinant Rat Activin-A $50/2µg $130/10µg $3,500/1mg Activin B, Active cyt-057 Recombinant Human Activin-B Active $50/2µg $130/10µg $5,200/1mg Activin B cyt-058 Recombinant Human Activin-B $50/2µg $130/10µg $4,800/1mg ACVR1 cyt-1140 Recombinant Human Activin A Receptor Type 1 $50/2µg $130/10µg $1,000/0.1mg ACVRL1 cyt-920 Recombinant Human Activin A Receptor Type II-Like 1 $50/2µg $130/10µg $5,200/1mg ACVR2A cyt-976 Recombinant Human Activin A Receptor Type 2A $50/2µg $130/10µg $5,200/1mg Acrp30 cyt-024 Human Adiponectin $50/2µg $130/10µg $1,000/0.1mg Acrp30 cyt-280 Recombinant Human Adiponectin $50/5µg $130/25µg $2,700/1mg Acrp30, His cyt-433 Recombinant Human Adiponectin, His tag $50/10µg $130/50µg $1,900/1mg Acrp30 (108-244) cyt-073 Recombinant Human Adiponectin (108-244 a.a.) $50/5µg $130/25µg $2,700/1mg Acrp30, HEK cyt-434 Recombinant Human Adiponectin glycosilated, HEK $50/2µg $130/10µg $4,680/1mg Acrp30, HMW cyt-764 Recombinant Human Adiponectin glycosilated, HMW Rich $50/2µg $130/10µg -

Prospec Product Catalog 2019
ProSpec Product Catalog 2019 QUALITY PROTEINS for scientific discoveries WE'VE GOT CHEMISTRY BRIGHTEN YOUR RESEARCH part for life sciences 6,000 proteins at your fingertips List by Molecule Name List by Molecule Name 제품별 자세한 설명은 www.prospecbio.com을 방문하세요. 부가세 별도 Molecule 제품번호 제품명 Size A / 가격(원) Size B / 가격(원) Size C / 가격(원) Name 1F8 Chagas cch-002 Recombinant 1F8 Chagas 5µg / 90,500 20µg / 199,000 1mg / 5,110,500 4-1BBL cyt-649 Recombinant Human 4-1BB Ligand, His Tag 5µg / 112,200 20µg / 220,800 1mg / 3,793,200 4-1BBL cyt-149 Recombinant Human 4-1BB Ligand 5µg / 90,500 20µg / 199,000 1mg / 3,771,400 4-1BBR cyt-916 Recombinant Mouse 4-1BB Receptor 1µg / 112,200 5µg / 220,800 50µg / 1,701,000 4-1BBR cyt-463 Recombinant Human 4-1BB Receptor 5µg / 90,500 20µg / 199,000 1mg / 3,771,400 4-1BBR cyt-137 Recombinant Human 4-1BB Receptor His Tag 5µg / 112,200 20µg / 220,800 1mg / 5,016,800 4-1BBR cyt-931 Recombinant Human 4-1BB Receptor sf9 2µg / 112,200 10µg / 220,800 1mg / 7,227,300 A2LD1 pro-172 Recombinant Human AIG2-Like Domain 1 2µg / 112,200 10µg / 220,800 1mg / 7,227,300 A2M pro-551 Human Alpha-2 Macroglobulin Protein 200µg / 90,500 1mg / 317,500 10mg / 2,389,800 Recombinant Human Alpha & Gamma-Adaptin Binding AAGAB pro-1479 5µg / 112,200 20µg / 220,800 1mg / 3,793,200 Protein Recombinant Human Adipogenesis Associated, Mth938 AAMDC pro-2116 5µg / 112,200 20µg / 220,800 1mg / 3,793,200 Domain Containing AARS enz-305 Recombinant Human Alanyl t-RNA Synthetase 5µg / 112,200 20µg / 220,800 1mg / 5,372,000 Recombinant Human Aminoadipate-Semialdehyde -

Table 4.3 Enzyme Activities and Glycerol Utilization and Ethanol Synthesis Fluxes for Wild-Type MG1655 and Strains Overexpressing Glycerol Utilization Enzymes
Abstract Understanding Fermentative Glycerol Metabolism and its Application for the Production of Fuels and Chemicals by James M. Clomburg Due to its availability, low-price, and higher degree of reduction than lignocellulosic sugars, glycerol has become an attractive carbon source for the production of fuels and reduced chemicals. However, this high degree of reduction of carbon atoms in glycerol also results in significant challenges in regard to its utilization under fermentative conditions. Therefore, in order to unlock the full potential of microorganisms for the fermentative conversion of glycerol into fuels and chemicals, a detailed understanding of the anaerobic fermentation of glycerol is required. The work presented here highlights a comprehensive experimental investigation into fermentative glycerol metabolism in Escherichia coli, which has elucidated several key pathways and mechanisms. The activity of both the fermentative and respiratory glycerol dissimilation pathways was found to be important for maximum glycerol utilization, a consequence of the metabolic cycle and downstream effects created by the essential involvement of PEP-dependent dihydroxyacetone kinase (DHAK) in the fermentative glycerol dissimilation pathway. The decoupling of this cycle is of central importance during fermentative glycerol metabolism, and while multiple decoupling mechanisms were identified, their relative inefficiencies dictated not only their level of involvement, but also iii implicated the activity of other pathways/enzymes, including fumarate reductase and pyruvate kinase. The central role of the PEP-dependent DHAK, an enzyme whose transcription was found to be regulated by the cyclic adenosine monophosphate (cAMP) receptor protein (CRP)-cAMP complex, was also tied to the importance of multiple fructose 1,6-bisphosphotases (FBPases) encoded by fbp, glpX, and yggF. -

Membrane Bound Dehydrogenase Enzymes and Related Electron Transport Systems in Acetobacter Aceti Var
Western Michigan University ScholarWorks at WMU Master's Theses Graduate College 10-1968 Membrane Bound Dehydrogenase Enzymes and Related Electron Transport Systems in Acetobacter Aceti Var. Liquefaciens: Their Induction and Pleiotropic Nature John C. Rakoczy Follow this and additional works at: https://scholarworks.wmich.edu/masters_theses Part of the Biochemistry Commons Recommended Citation Rakoczy, John C., "Membrane Bound Dehydrogenase Enzymes and Related Electron Transport Systems in Acetobacter Aceti Var. Liquefaciens: Their Induction and Pleiotropic Nature" (1968). Master's Theses. 3214. https://scholarworks.wmich.edu/masters_theses/3214 This Masters Thesis-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Master's Theses by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. MEMBRANE BOUND DEHYDROGENASE ENZYMES AND RELATED ELECTRON TRANSPORT SYSTEMS IN ACETOBACTER ACETI VAR. LIQUEFACIENS: THEIR INDUCTION AND PLEIOTROPIC NATURE by John C. Rakoczy A Thesis Submitted to the Faculty of the School of Graduate Studies in partial fulfillment of the Degree of Master of Arts Western Michigan University Kalamazooj Michigan October 1968 " Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGEMENTS The author wishes to express his gratitude to Dr. Stephen B. Friedman for his patience, guidance, and encouragement throughout the present study, and to Dr. Robert C. Eisenberg for his kindly advice and assis tance. Sincere appreciation is extended to the many persons, too numerous to mention, who have given their time, advice and encouragement to the enterprise. John C. -

Evolution of D-Lactate Dehydrogenase Activity from Glycerol Dehydrogenase and Its Utility for D-Lactate Production from Lignocellulose
Evolution of D-lactate dehydrogenase activity from glycerol dehydrogenase and its utility for D-lactate production from lignocellulose Qingzhao Wang, Lonnie O. Ingram, and K. T. Shanmugam1 Department of Microbiology and Cell Science, University of Florida, Gainesville, FL 32611 Edited by Arnold L. Demain, Drew University, Madison, NJ, and approved October 6, 2011 (received for review July 9, 2011) Lactic acid, an attractive, renewable chemical for production of from glucose and sucrose at temperatures between 30 °C and biobased plastics (polylactic acid, PLA), is currently commercially 40 °C (8, 10). Derivatives of Escherichia coli are the only known produced from food-based sources of sugar. Pure optical isomers commercial D(−)-lactic acid producers (11, 12) and these also of lactate needed for PLA are typically produced by microbial fer- operate optimally at 40 °C or lower. mentation of sugars at temperatures below 40 °C. Bacillus coagu- Alternative sources of fermentable sugars such as lignocellu- lans produces L(+)-lactate as a primary fermentation product and losic biomass and improved microbial biocatalysts are needed grows optimally at 50 °C and pH 5, conditions that are optimal to eliminate the use of food carbohydrates (glucose, sucrose) for for activity of commercial fungal cellulases. This strain was engi- lactic acid production (13). With cellulose as a feedstock, how- neered to produce D(−)-lactate by deleting the native ldh (L-lactate ever, commercial fungal cellulases represent a significant process dehydrogenase) and alsS (acetolactate synthase) genes to impede cost. This cost could be reduced by the development of thermo- anaerobic growth, followed by growth-based selection to isolate tolerant biocatalysts that effectively ferment under conditions suppressor mutants that restored growth. -

2019 Strain Engineering for Mi
Biotechnology Advances 37 (2019) 538–568 Contents lists available at ScienceDirect Biotechnology Advances journal homepage: www.elsevier.com/locate/biotechadv Research review paper Strain engineering for microbial production of value-added chemicals and T fuels from glycerol ⁎ Adam W. Westbrook , Dragan Miscevic, Shane Kilpatrick, Mark R. Bruder, Murray Moo-Young, ⁎ C. Perry Chou Department of Chemical Engineering, Waterloo, Ontario, Canada ARTICLE INFO ABSTRACT Keyword: While the widespread reliance on fossil fuels is driven by their low cost and relative abundance, this fossil-based Glycerol economy has been deemed unsustainable and, therefore, the adoption of sustainable and environmentally Metabolic engineering compatible energy sources is on the horizon. Biorefinery is an emerging approach that integrates metabolic Biorefinery engineering, synthetic biology, and systems biology principles for the development of whole-cell catalytic Biofuels platforms for biomanufacturing. Due to the high degree of reduction and low cost, glycerol, either refined or E. coli crude, has been recognized as an ideal feedstock for the production of value-added biologicals, though microbial Clostridium Klebsiella dissimilation of glycerol sometimes can be difficult particularly under anaerobic conditions. While strain de- Citrobacter velopment for glycerol biorefinery is widely reported in the literature, few, if any, commercialized bioprocesses Lactobacillus have been developed as a result, such that engineering of glycerol metabolism in microbial hosts remains an untapped opportunity in biomanufacturing. Here we review the recent progress made in engineering microbial hosts for the production of biofuels, diols, organic acids, biopolymers, and specialty chemicals from glycerol. We begin with a broad outline of the major pathways for fermentative and respiratory glycerol dissimilation and key end metabolites, and then focus our analysis on four key genera of bacteria known to naturally dissimilate glycerol, i.e.