Insights Into an Alternative Pathway for Glycerol Metabolism in a Glycerol Kinase Deficient
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

I HIGH MASS ACCURACY COUPLED to SPATIALLY-DIRECTED
HIGH MASS ACCURACY COUPLED TO SPATIALLY-DIRECTED PROTEOMICS FOR IMPROVED PROTEIN IDENTIFICATIONS IN IMAGING MASS SPECTROMETRY EXPERIMENTS By David Geoffrey Rizzo Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Chemistry August, 2016 Nashville, Tennessee Approved: Richard M. Caprioli, Ph.D. Kevin L. Schey, Ph.D. John A. McLean, Ph.D. Michael P. Stone, Ph.D. i Copyright © 2016 by David Geoffrey Rizzo All Rights Reserved ii This work is dedicated to my family and friends, who have shown nothing but support for me in all of life’s endeavors. iii ACKNOWLEDGEMENTS “As we express our gratitude, we must never forget that the highest appreciation is not to utter words, but to live by them.” - John F. Kennedy – There are many people I must thank for showing kindness, encouragement, and support for me during my tenure as a graduate student. First and foremost, I would like to thank my research advisor, Richard Caprioli, for providing both ample resources and guidance that allowed me to grow as a scientist. Our discussions about my research and science in general have helped me become a much more focused and discerning analytical chemist. I must also thank my Ph.D. committee members, Drs. Kevin Schey, John McLean, and Michael Stone, who have brought valuable insight into my research and provided direction along the way. My undergraduate advisor, Dr. Facundo Fernández, encouraged me to begin research in his lab and introduced me to the world of mass spectrometry. -

Glycerol Dehydrogenase from Gluconobacter Industrius
Agric. Biol Chem., 49 (4), 1001 -1010, 1985 1001 Solubilization, Purification and Properties of Membrane-bound Glycerol Dehydrogenase from Gluconobacter industrius Minoru Ameyama,Emiko Shinagawa, Kazunobu Matsushita and Osao Adachi Laboratory of Applied Microbiology, Department of Agricultural Chemistry, Faculty of Agriculture, Yamaguchi University, Yamaguchi 753, Japan Received July 30, 1984 Membrane-bound glycerol dehydrogenase was solubilized and purified about 100-fold from the membraneof Gluconobacter industrius IFO 3260 grown on a glycerol-glutamate medium. Solubilization of the enzyme was successfully achieved by use of 0.5% dimethyldodecylamineoxide in 0.05 m Tris-HCl, pH 8.0. Alcohol dehydrogenase and D-glucose dehydrogenase, which were abundantly formed in the same bacterial membrane, were eliminated on solubilization. Glycerol dehydrogenase was further purified through fractionation with polyethylene glycol 6000. The enzymeshowed a broad substrate specificity and various kinds of polyhydroxyl alcohols, in addition to glycerol, were rapidly oxidized in the presence of 2,6-dichlorophenolindophenoi and phenazine methosulfate as the electron acceptor but NADand NADPwere inert. The enzyme was proved to be a quinoprotein in which pyrroloquinoline quinone functioned as the prosthetic group. The first report on microbial oxidation of localization of the oxidase system in cells of G. glycerol to dihydroxyacetone was by Bertrand liquefaciens and found that the oxidation of with a strain capable of L-sorbose fermen- glycerol and raeso-erythritol -

Negative Regulation of Diacylglycerol Kinase &Theta
Cell Death and Differentiation (2010) 17, 1059–1068 & 2010 Macmillan Publishers Limited All rights reserved 1350-9047/10 $32.00 www.nature.com/cdd Negative regulation of diacylglycerol kinase h mediates adenosine-dependent hepatocyte preconditioning G Baldanzi1,5, E Alchera2,5, C Imarisio2, M Gaggianesi1, C Dal Ponte2, M Nitti3, C Domenicotti3, WJ van Blitterswijk4, E Albano2, A Graziani1,5 and R Carini*,2,5 In liver ischemic preconditioning (IP), stimulation of adenosine A2a receptors (A2aR) prevents ischemia/reperfusion injury by promoting diacylglycerol-mediated activation of protein kinase C (PKC). By concerting diacylglycerol to phosphatidic acid, diacylglycerol kinases (DGKs) act as terminator of diacylglycerol signalling. This study investigates the role of DGK in the development of hepatocyte IP. DGK activity and cell viability were evaluated in isolated rat hepatocytes preconditioned by 10 min hypoxia followed by 10 min re-oxygenation or by the treatment with the A2aR agonist, CGS21680, and subsequently exposed to prolonged hypoxia. We observed that after IP or A2aR activation, a decrease in DGK activity was associated with the onset of hepatocyte tolerance to hypoxia. CGS21680-induced stimulation of A2aR specifically inhibited DGK isoform h by activating RhoA–GTPase. Consistently, both siRNA-mediated downregulation of DGK h and hepatocyte pretreatment with the DGK inhibitor R59949 induced cell tolerance to hypoxia. The pharmacological inhibition of DGK was associated with the diacylglycerol- dependent activation of PKC d and e and of their downstream target p38 MAPK. In conclusion, we unveil a novel signalling pathway contributing to the onset of hepatocyte preconditioning, which through RhoA–GTPase, couples A2aR to the downregulation of DGK. -

Supplementary Materials
Supplementary Materials Figure S1. Differentially abundant spots between the mid-log phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 3. Figure S2. Differentially abundant spots between the stationary phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 4. S2 Table S1. Summary of the non-polysaccharide degrading proteins identified in the B. proteoclasticus cytosol by 2DE/MALDI-TOF. Protein Locus Location Score pI kDa Pep. Cov. Amino Acid Biosynthesis Acetylornithine aminotransferase, ArgD Bpr_I1809 C 1.7 × 10−4 5.1 43.9 11 34% Aspartate/tyrosine/aromatic aminotransferase Bpr_I2631 C 3.0 × 10−14 4.7 43.8 15 46% Aspartate-semialdehyde dehydrogenase, Asd Bpr_I1664 C 7.6 × 10−18 5.5 40.1 17 50% Branched-chain amino acid aminotransferase, IlvE Bpr_I1650 C 2.4 × 10−12 5.2 39.2 13 32% Cysteine synthase, CysK Bpr_I1089 C 1.9 × 10−13 5.0 32.3 18 72% Diaminopimelate dehydrogenase Bpr_I0298 C 9.6 × 10−16 5.6 35.8 16 49% Dihydrodipicolinate reductase, DapB Bpr_I2453 C 2.7 × 10−6 4.9 27.0 9 46% Glu/Leu/Phe/Val dehydrogenase Bpr_I2129 C 1.2 × 10−30 5.4 48.6 31 64% Imidazole glycerol phosphate synthase Bpr_I1240 C 8.0 × 10−3 4.7 22.5 8 44% glutamine amidotransferase subunit Ketol-acid reductoisomerase, IlvC Bpr_I1657 C 3.8 × 10−16 -

Promiscuity in the Part-Phosphorylative Entner–Doudoroff Pathway of the Archaeon Sulfolobus Solfataricus
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS 30191 FEBS Letters 579 (2005) 6865–6869 Promiscuity in the part-phosphorylative Entner–Doudoroff pathway of the archaeon Sulfolobus solfataricus Henry J. Lamblea, Alex Theodossisb, Christine C. Milburnb, Garry L. Taylorb, Steven D. Bullc, David W. Hougha, Michael J. Dansona,* a Centre for Extremophile Research, Department of Biology and Biochemistry, University of Bath, Bath BA2 7AY, UK b Centre for Biomolecular Sciences, University of St. Andrews, North Haugh, St. Andrews, Fife KY16 9ST, UK c Department of Chemistry, University of Bath, Bath BA2 7AY, UK Received 19 September 2005; revised 3 November 2005; accepted 3 November 2005 Available online 1 December 2005 Edited by Stuart Ferguson dation of both glucose and galactose, producing gluconate or Abstract The hyperthermophilic archaeon Sulfolobus solfatari- cus metabolises glucose and galactose by a ÔpromiscuousÕ non- galactonate, respectively [6]. Gluconate dehydratase then phosphorylative variant of the Entner–Doudoroff pathway, in catalyses the dehydration of gluconate to D-2-keto-3-deoxyg- which a series of enzymes have sufficient substrate promiscuity luconate (KDG) and galactonate to D-2-keto-3-deoxygalacto- to permit the metabolism of both sugars. Recently, it has been nate (KDGal) [7]. Both these compounds are cleaved by KDG proposed that the part-phosphorylative Entner–Doudoroff path- aldolase to yield pyruvate and glyceraldehyde [6]. Glyceralde- way occurs in parallel in S. solfataricus as an alternative route hyde dehydrogenase is then thought to oxidise glyceraldehyde for glucose metabolism. In this report we demonstrate, by to glycerate, which is phosphorylated by glycerate kinase to in vitro kinetic studies of D-2-keto-3-deoxygluconate (KDG) ki- give 2-phosphoglycerate. -

Table S1. List of Oligonucleotide Primers Used
Table S1. List of oligonucleotide primers used. Cla4 LF-5' GTAGGATCCGCTCTGTCAAGCCTCCGACC M629Arev CCTCCCTCCATGTACTCcgcGATGACCCAgAGCTCGTTG M629Afwd CAACGAGCTcTGGGTCATCgcgGAGTACATGGAGGGAGG LF-3' GTAGGCCATCTAGGCCGCAATCTCGTCAAGTAAAGTCG RF-5' GTAGGCCTGAGTGGCCCGAGATTGCAACGTGTAACC RF-3' GTAGGATCCCGTACGCTGCGATCGCTTGC Ukc1 LF-5' GCAATATTATGTCTACTTTGAGCG M398Arev CCGCCGGGCAAgAAtTCcgcGAGAAGGTACAGATACGc M398Afwd gCGTATCTGTACCTTCTCgcgGAaTTcTTGCCCGGCGG LF-3' GAGGCCATCTAGGCCATTTACGATGGCAGACAAAGG RF-5' GTGGCCTGAGTGGCCATTGGTTTGGGCGAATGGC RF-3' GCAATATTCGTACGTCAACAGCGCG Nrc2 LF-5' GCAATATTTCGAAAAGGGTCGTTCC M454Grev GCCACCCATGCAGTAcTCgccGCAGAGGTAGAGGTAATC M454Gfwd GATTACCTCTACCTCTGCggcGAgTACTGCATGGGTGGC LF-3' GAGGCCATCTAGGCCGACGAGTGAAGCTTTCGAGCG RF-5' GAGGCCTGAGTGGCCTAAGCATCTTGGCTTCTGC RF-3' GCAATATTCGGTCAACGCTTTTCAGATACC Ipl1 LF-5' GTCAATATTCTACTTTGTGAAGACGCTGC M629Arev GCTCCCCACGACCAGCgAATTCGATagcGAGGAAGACTCGGCCCTCATC M629Afwd GATGAGGGCCGAGTCTTCCTCgctATCGAATTcGCTGGTCGTGGGGAGC LF-3' TGAGGCCATCTAGGCCGGTGCCTTAGATTCCGTATAGC RF-5' CATGGCCTGAGTGGCCGATTCTTCTTCTGTCATCGAC RF-3' GACAATATTGCTGACCTTGTCTACTTGG Ire1 LF-5' GCAATATTAAAGCACAACTCAACGC D1014Arev CCGTAGCCAAGCACCTCGgCCGAtATcGTGAGCGAAG D1014Afwd CTTCGCTCACgATaTCGGcCGAGGTGCTTGGCTACGG LF-3' GAGGCCATCTAGGCCAACTGGGCAAAGGAGATGGA RF-5' GAGGCCTGAGTGGCCGTGCGCCTGTGTATCTCTTTG RF-3' GCAATATTGGCCATCTGAGGGCTGAC Kin28 LF-5' GACAATATTCATCTTTCACCCTTCCAAAG L94Arev TGATGAGTGCTTCTAGATTGGTGTCggcGAAcTCgAGCACCAGGTTG L94Afwd CAACCTGGTGCTcGAgTTCgccGACACCAATCTAGAAGCACTCATCA LF-3' TGAGGCCATCTAGGCCCACAGAGATCCGCTTTAATGC RF-5' CATGGCCTGAGTGGCCAGGGCTAGTACGACCTCG -

Domains, Amino Acid Residues, and New Isoforms of Caenorhabditis Elegans Diacylglycerol Kinase 1 (DGK-1) Important for Terminating Diacylglycerol Signaling in Vivo*□S
Supplemental Material can be found at: http://www.jbc.org/content/suppl/2004/12/06/M409460200.DC1.html THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 280, No. 4, Issue of January 28, pp. 2730–2736, 2005 © 2005 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. Domains, Amino Acid Residues, and New Isoforms of Caenorhabditis elegans Diacylglycerol Kinase 1 (DGK-1) Important for Terminating Diacylglycerol Signaling in Vivo*□S Received for publication, August 17, 2004, and in revised form, November 22, 2004 Published, JBC Papers in Press, November 24, 2004, DOI 10.1074/jbc.M409460200 Antony M. Jose‡ and Michael R. Koelle§¶ From the ‡Departments of Molecular, Cellular, and Developmental Biology and §Molecular Biophysics and Biochemistry, Yale University School of Medicine, New Haven, Connecticut 06520 Diacylglycerol kinases (DGKs) inhibit diacylglycerol numerous cellular processes mediated by neurotransmitters, (DAG) signaling by phosphorylating DAG. DGK-1, the growth factors, and hormones (3). DAG also activates the syn- Caenorhabditis elegans ortholog of human neuronal aptic vesicle priming protein UNC-13 (4, 5) to control neuro- DGK, inhibits neurotransmission to control behavior. transmission and certain transient receptor potential cation Downloaded from DGK-1, like DGK, has three cysteine-rich domains channels (6). In humans, nine DGK isozymes have been iden- (CRDs), a pleckstrin homology domain, and a kinase tified (DGK␣, , ␥, ␦, , ⑀, , , and ), but their physiological domain. To identify DGK domains and amino acid resi- functions remain largely unknown (1, 2). dues critical for terminating DAG signaling in vivo,we C. elegans DGK-1 provides a genetically tractable model for analyzed 20 dgk-1 mutants defective in DGK-1-con- elucidating the physiological functions of diacylglycerol ki- trolled behaviors. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
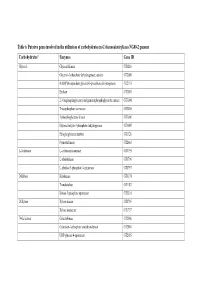
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

Coupling of Creatine Kinase to Glycolytic Enzymes at the Sarcomeric I-Band of Skeletal Muscle: a Biochemical Study in Situ
Journal of Muscle Research and Cell Motility 21: 691±703, 2000. 691 Ó 2000 Kluwer Academic Publishers. Printed in the Netherlands. Coupling of creatine kinase to glycolytic enzymes at the sarcomeric I-band of skeletal muscle: a biochemical study in situ THERESIA KRAFT2,*, T. HORNEMANN1, M. STOLZ1,à, V. NIER2, and T. WALLIMANN1 1Swiss Federal Institute of Technology, Institute of Cell Biology, ETH ZuÈrich, HoÈnggerberg, CH-8093 ZuÈrich, Switzerland; 2Medizinische Hochschule Hannover, Molekular- und Zellphysiologie, D-30625 Hannover, Germany Received 29 September 2000; accepted in revised form 3 October 2000 Abstract The speci®c interaction of muscle type creatine-kinase (MM-CK) with the myo®brillar M-line was demonstrated by exchanging endogenous MM-CK with an excess of ¯uorescently labeled MM-CK in situ, using chemically skinned skeletal muscle ®bers and confocal microscopy. No binding of labeled MM-CK was noticed at the I-band of skinned ®bers, where the enzyme is additionally located in vivo, as shown earlier by immuno¯uorescence staining of cryosections of intact muscle. However, when rhodamine-labeled MM-CK was diused into skinned ®bers that had been preincubated with phosphofructokinase (PFK), a glycolytic enzyme known to bind to actin, a striking in vivo- like interaction of Rh-MM-CK with the I-band was found, presumably mediated by binding of Rh-MM-CK to the glycolytic enzyme. Aldolase, another actin-binding glycolytic enzyme was also able to bind Rh-MM-CK to the I- band, but formation of the complex occurred preferably at long sarcomere length (>3.0 lm). Neither pyruvate kinase, although known for its binding to actin, nor phosphoglycerate kinase (PGK), not directly interacting with the I-band itself, did mediate I-band targeting of MM-CK. -

Prefoldin-Like Bud27 Influences the Transcription of Ribosomal Components and Ribosome Biogenesis in Saccharomyces Cerevisiae
Downloaded from rnajournal.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press Prefoldin-like Bud27 influences the transcription of ribosomal components and ribosome biogenesis in Saccharomyces cerevisiae VERÓNICA MARTÍNEZ-FERNÁNDEZ,1,7,8 ABEL CUEVAS-BERMÚDEZ,1,8 FRANCISCO GUTIÉRREZ-SANTIAGO,1,8 ANA I. GARRIDO-GODINO,1 OLGA RODRÍGUEZ-GALÁN,2,3 ANTONIO JORDÁN-PLA,4 SERGIO LOIS,5 JUAN C. TRIVIÑO,5 JESÚS DE LA CRUZ,2,3 and FRANCISCO NAVARRO1,6 1Departamento de Biología Experimental-Genética, Universidad de Jaén, Paraje de las Lagunillas, s/n, E-23071, Jaén, Spain 2Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, E-41013 Seville, Spain 3Departamento de Genética, Universidad de Sevilla, E-41012 Seville, Spain 4ERI Biotecmed, Facultad de Biológicas, Universitat de València, E-46100 Burjassot, Valencia, Spain 5Sistemas Genómicos. Ronda de Guglielmo Marconi, 6, 46980 Paterna, Valencia, Spain 6Centro de Estudios Avanzados en Aceite de Oliva y Olivar, Universidad de Jaén, Paraje de las Lagunillas, s/n, E-23071, Jaén, Spain ABSTRACT Understanding the functional connection that occurs for the three nuclear RNA polymerases to synthesize ribosome com- ponents during the ribosome biogenesis process has been the focal point of extensive research. To preserve correct ho- meostasis on the production of ribosomal components, cells might require the existence of proteins that target a common subunit of these RNA polymerases to impact their respective activities. This work describes how the yeast prefoldin-like Bud27 protein, which physically interacts with the Rpb5 common subunit of the three RNA polymerases, is able to mod- ulate the transcription mediated by the RNA polymerase I, likely by influencing transcription elongation, the transcription of the RNA polymerase III, and the processing of ribosomal RNA.