Fasturtec, INN-Rasburicase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
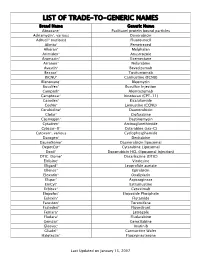
Trade-To-Generic Names
LIST OF TRADE-TO-GENERIC NAMES Brand Name Generic Name Abraxane® Paclitaxel protein bound particles Adriamycin®, various Doxorubicin Adrucil® (various) Fluorouracil Alimta® Pemetrexed Alkeran® Melphalan Arimidex® Anastrozole Aromasin® Exemestane Arranon® Nelarabine Avastin® Bevacizumab Bexxar® Tositumomab BiCNU® Carmustine (BCNU) Blenoxane® Bleomycin Busulfex® Busulfan Injection Campath® Alemtuzumab Camptosar® Irinotecan (CPT-11) Casodex® Bicalutamide CeeNu® Lomustine (CCNU) Cerubidine® Daunorubicin Clolar® Clofarabine Cosmegen® Dactinomycin Cytadren® Aminoglutethimide Cytosar-U® Cytarabine (ara-C) Cytoxan®, various Cyclophosphamide Dacogen® Decitabine DaunoXome® Daunorubicin liposomal DepotCyt® Cytarabine Liposomal Doxil® Doxorubicin HCL (liposomal injection) DTIC-Dome® Dacarbazine (DTIC) Eldisine® Vindesine Eligard® Leuprolide acetate Ellence® Epirubicin Eloxatin® Oxaliplatin Elspar® Asparaginase EmCyt® Estramustine Erbitux® Cetuximab Etopofos® Etoposide Phosphate Eulexin® Flutamide Fareston® Toremifene Faslodex® Fluvestrant Femara® Letrozole Fludara® Fludarabine Gemzar® Gemcitabine Gleevec® Imatinib Gliadel® Carmustine Wafer Halotestin® Fluoxymesterone Last Updated on January 15, 2007 Brand Name Generic Name Herceptin® Trastuzumab Hexalen® Altretamine Hycamtin® Topotecan Hydrea® Hydroxyurea Idamycin® Idarubicin Ifex® Ifosfamide Intron A® Interferon alfa-2b Iressa® Gefitinib Leukeran® Chlorambucil Leukine® Sargramostim Leustatin® Cladribine Lupron depot® Leuprolide acetate depot Lupron® Leuprolide acetate Matulane® Procarbazine Megace® -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

BC Cancer Protocol Summary for Treatment of Lymphoma with Dose- Adjusted Etoposide, Doxorubicin, Vincristine, Cyclophosphamide
BC Cancer Protocol Summary for Treatment of Lymphoma with Dose- Adjusted Etoposide, DOXOrubicin, vinCRIStine, Cyclophosphamide, predniSONE and riTUXimab with Intrathecal Methotrexate Protocol Code LYEPOCHR Tumour Group Lymphoma Contact Physician Dr. Laurie Sehn Dr. Kerry Savage ELIGIBILITY: One of the following lymphomas: . Patients with an aggressive B-cell lymphoma and the presence of a dual translocation of MYC and BCL2 (i.e., double-hit lymphoma). Histologies may include DLBCL, transformed lymphoma, unclassifiable lymphoma, and intermediate grade lymphoma, not otherwise specified (NOS). Patients with Burkitt lymphoma, who are not candidates for CODOXM/IVACR (such as those over the age of 65 years, or with significant co-morbidities) . Primary mediastinal B-cell lymphoma Ensure patient has central line EXCLUSIONS: . Cardiac dysfunction that would preclude the use of an anthracycline. TESTS: . Baseline (required before first treatment): CBC and diff, platelets, BUN, creatinine, bilirubin. ALT, LDH, uric acid . Baseline (required, but results do not have to be available to proceed with first treatment): results must be checked before proceeding with cycle 2): HBsAg, HBcoreAb, . Baseline (optional, results do not have to be available to proceed with first treatment): HCAb, HIV . Day 1 of each cycle: CBC and diff, platelets, (and serum bilirubin if elevated at baseline; serum bilirubin does not need to be requested before each treatment, after it has returned to normal), urinalysis for microscopic hematuria (optional) . Days 2 and 5 of each cycle (or days of intrathecal treatment): CBC and diff, platelets, PTT, INR . For patients on cyclophosphamide doses greater than 2000 mg: Daily urine dipstick for blood starting on day cyclophosphamide is given. -

Rasburicase (Elitek)
Rasburicase (Elitek) ELITEK™ (rasburicase) BOXED WARNINGS Anaphylaxis ELITEK may cause severe hypersensitivity reactions including anaphylaxis. ELITEK should be immediately and permanently discontinued in any patient developing clinical evidence of a serious hypersensitivity reaction (see WARNINGS, Anaphylaxis and ADVERSE REACTIONS, Immunogenicity). Hemolysis ELITEK administered to patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency can cause severe hemolysis. ELITEK administration should be immediately and permanently discontinued in any patient developing hemolysis. It is recommended that patients at higher risk for G6PD deficiency (e.g., patients of African or Mediterranean ancestry) be screened prior to starting ELITEK therapy (see CONTRAINDICATIONS and WARNINGS, Hemolysis). Methemoglobinemia ELITEK use has been associated with Methemoglobinemia. ELITEK administration should be immediately and permanently discontinued in any patient identified as having developed methemoglobinemia (see WARNINGS, Methemoglobinemia). Interference with Uric Acid Measurements ELITEK will cause enzymatic degradation of the uric acid within blood samples left at room temperature, resulting in spuriously low uric acid levels. To ensure accurate measurements, blood must be collected into pre-chilled tubes containing heparin anticoagulent and immediately immersed and maintained in an ice water bath; plasma samples must be assayed within 4 hours of sample collection (see PRECAUTIONS, Laboratory Test Interactions). DESCRIPTION ELITEK (rasburicase) is a recombinant urate-oxidase enzyme produced by a genetically modified Saccharomyces cerevisiae strain. The cDNA coding for rasburicase was cloned from a strain of Aspergillus flavus. Rasburicase is a tetrameric protein with identical subunits of a molecular mass of about 34 kDa. The molecular formula of the monomer is C1523 H2383 N417 O462 S7. The monomer, made up of a single 301 amino acid polypeptide chain, has no intra- or inter-disulfide bridges and is N-terminal acetylated. -

Approved Cancer Drugs for Children
U.S. FOOD & DRUG li1 ADMINISTRATION Approved Cancer Drugs for Children Amy Barone, MD, MSCI March 15, 2019 Frequent Criticism: Too few drugs approved for pediatric cancer “Since 1980, only 4 drugs have been approved for the first instance for use in children.” - Coalition Against Childhood Cancer “In the last 20 years, only two new drugs have been approved that were specifically developed to treat children with cancer.” – St. Baldricks “Over the past 20 years, the FDA has approved about 190 new cancer treatments for adults but only three for children.” USA Today “Since 1980, fewer than 10 drugs have been developed for use in children with cancer. Only three drugs have been approved for use in children. Only four additional new drugs have been approved for use by both adults and children.” - National Pediatric Cancer Foundation “15 oncology drugs were approved by the FDA for pediatric use between 1948 and 2003.” – Managed Care “From 1980 to 2017, only 11 drugs (already approved in adults) have been approved to use in children with cancer” - Coalition Against Childhood Cancer 2 Question: How many drugs are FDA approved to treat pediatric cancer? • A: 11 • B: 34 • C: 4 • D: 15 3 “There’s no tragedy in life like the death of a child.” - Dwight D. Eisenhower 4 Antitoxin Contamination • Early 1900s – Animal anti-sera given to patients with cholera, typhoid, etc. • A Horse named “Jim” – Contaminated serum – Anti-toxin resulted in deaths of 13 children • Second incident – Contaminated smallpox vaccine killed 9 children Laws Enacted 1902 – Biologics Control Act 1906 – Pure Food and Drug Act 6 Elixir Sulfanilamide Tragedy O ' 7 Law Enacted The Food, Drug and Cosmetic (FDC) Act of 1938 8 Thalidomide T~~:••~ ~ . -

Estonian Statistics on Medicines 2016 1/41
Estonian Statistics on Medicines 2016 ATC code ATC group / Active substance (rout of admin.) Quantity sold Unit DDD Unit DDD/1000/ day A ALIMENTARY TRACT AND METABOLISM 167,8985 A01 STOMATOLOGICAL PREPARATIONS 0,0738 A01A STOMATOLOGICAL PREPARATIONS 0,0738 A01AB Antiinfectives and antiseptics for local oral treatment 0,0738 A01AB09 Miconazole (O) 7088 g 0,2 g 0,0738 A01AB12 Hexetidine (O) 1951200 ml A01AB81 Neomycin+ Benzocaine (dental) 30200 pieces A01AB82 Demeclocycline+ Triamcinolone (dental) 680 g A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+ Thymol (dental) 3094 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+ Cetylpyridinium chloride (gingival) 227150 g A01AD81 Lidocaine+ Cetrimide (O) 30900 g A01AD82 Choline salicylate (O) 864720 pieces A01AD83 Lidocaine+ Chamomille extract (O) 370080 g A01AD90 Lidocaine+ Paraformaldehyde (dental) 405 g A02 DRUGS FOR ACID RELATED DISORDERS 47,1312 A02A ANTACIDS 1,0133 Combinations and complexes of aluminium, calcium and A02AD 1,0133 magnesium compounds A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 811120 pieces 10 pieces 0,1689 A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 3101974 ml 50 ml 0,1292 A02AD83 Calcium carbonate+ Magnesium carbonate (O) 3434232 pieces 10 pieces 0,7152 DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 46,1179 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 2,3855 A02BA02 Ranitidine (O) 340327,5 g 0,3 g 2,3624 A02BA02 Ranitidine (P) 3318,25 g 0,3 g 0,0230 A02BC Proton pump inhibitors 43,7324 A02BC01 Omeprazole -

Chapter 4: Tumor Lysis Syndrome
Chapter 4: Tumor Lysis Syndrome † Amaka Edeani, MD,* and Anushree Shirali, MD *Kidney Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland; and †Section of Nephrology, Yale University School of Medicine, New Haven, Connecticut INTRODUCTION class specifies patients with normal laboratory and clinical parameters as having no LTLS or CTLS. Tumor lysis syndrome (TLS) is a constellation of Cairo and Bishop also proposed a grading system metabolic abnormalities resulting from either spon- combiningthedefinitionsofnoTLS,LTLS,andCTLS, taneous or chemotherapy-induced tumor cell death. with the maximal clinical manifestations in each Tumor cytotoxicity releases intracellular contents, affectedorgandefiningthegrade ofTLS(1).Although including nucleic acids, proteins, and electrolytes this grading system attempts to provide uniform def- into the systemic circulation and may lead to devel- initions to TLS severity, it is not widely used in clinical opment of hyperuricemia, hyperphosphatemia, hy- practice. pocalcemia,andhyperkalemia.Clinically,thisresults The Cairo-Bishop classification is not immune to in multiorgan effects such as AKI, cardiac arrhyth- critique despite its common use. Specifically, patients mias, and seizures (1,2). TLS is the most common with TLS may not always have two or more abnor- oncologic emergency (3), and without prompt rec- malities present at once, but one metabolic derange- ognition and early therapeutic intervention, mor- ment may precede another (2). Furthermore, a 25% bidity and mortality is high. change from baseline may not always be significant if it does not result in a value outside the normal range (2). From a renal standpoint, Wilson and Berns (5) DEFINITION have noted that defining AKI on the basis of a creat- inine value .1.5 times the upper limit of normal Hande and Garrow (4) first initiated a definition of does not clearly distinguish CKD from AKI. -

Nebraska Medicine IV Push Medication Review
Nebraska Medicine IV Push Medication Review Adults Adults Pediatrics Pediatrics Can Med be Chemotherapy or If Med can be administered Can Med be If Med can be administered Drug Name administered IV Mab? IV push, how fast can it be administered IV IV push, how fast can it be push or < 5 pushed? push in? (Yes/No) pushed? min? (Yes/No) acetaZOLAMIDE No Yes 500 mg over 3 min Yes 500 mg over 3 min acetylcysteine No No No acyclovir No No No alteplase PE No No No alteplase stroke No No No amikacin No No No aminocaproic acid No No No aminophylline No No No amiodarone No No No ampho B (liposomal) No No No amphotericin B No No No ampicillin No Yes 500 mg over 3-5 min Yes 500 mg over 3-5 min ampicillin/sulbact No No No anthymocyte EQUINE No No No anthymocyte RABBI No No No anti-inhibitor FEIBA No No No antihem factor No Yes 5 min or less Yes 5 min or less antithrombin III No No No arginine No No No ascorbic acid No No No azithromycin No No No aztreonam No Yes 3 to 5 min Yes 3 to 5 min caffeine/sod benzoa No No No calcium chloride No No No calcium gluconate No No No ceFAZolin No Yes 3 to 5 min Yes 3 to 5 min cefePIME No Yes 5 min Yes 3 to 5 min cefoTAXime No Yes 3 to 5 min Yes 3 to 5 min cefOXitin No Yes 3 to 5 min Yes 3 to 5 min ceftaroline No No No cefTAZidime No Yes 3 to 5 min Yes 3 to 5 min cefTAZidime/avibacta No No No ceftolozane/tazobac No No No Nebraska Medicine IV Push Medication Review Adults Adults Pediatrics Pediatrics Can Med be Chemotherapy or If Med can be administered Can Med be If Med can be administered Drug Name administered IV -

Standard Oncology Criteria C16154-A
Prior Authorization Criteria Standard Oncology Criteria Policy Number: C16154-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DATE DUE BEFORE 03/2016 12/2/2020 1/26/2022 HCPCS CODING TYPE OF CRITERIA LAST P&T APPROVAL/VERSION N/A RxPA Q1 2021 20210127C16154-A PRODUCTS AFFECTED: See dosage forms DRUG CLASS: Antineoplastic ROUTE OF ADMINISTRATION: Variable per drug PLACE OF SERVICE: Retail Pharmacy, Specialty Pharmacy, Buy and Bill- please refer to specialty pharmacy list by drug AVAILABLE DOSAGE FORMS: Abraxane (paclitaxel protein-bound) Cabometyx (cabozantinib) Erwinaze (asparaginase) Actimmune (interferon gamma-1b) Calquence (acalbrutinib) Erwinia (chrysantemi) Adriamycin (doxorubicin) Campath (alemtuzumab) Ethyol (amifostine) Adrucil (fluorouracil) Camptosar (irinotecan) Etopophos (etoposide phosphate) Afinitor (everolimus) Caprelsa (vandetanib) Evomela (melphalan) Alecensa (alectinib) Casodex (bicalutamide) Fareston (toremifene) Alimta (pemetrexed disodium) Cerubidine (danorubicin) Farydak (panbinostat) Aliqopa (copanlisib) Clolar (clofarabine) Faslodex (fulvestrant) Alkeran (melphalan) Cometriq (cabozantinib) Femara (letrozole) Alunbrig (brigatinib) Copiktra (duvelisib) Firmagon (degarelix) Arimidex (anastrozole) Cosmegen (dactinomycin) Floxuridine Aromasin (exemestane) Cotellic (cobimetinib) Fludara (fludarbine) Arranon (nelarabine) Cyramza (ramucirumab) Folotyn (pralatrexate) Arzerra (ofatumumab) Cytosar-U (cytarabine) Fusilev (levoleucovorin) Asparlas (calaspargase pegol-mknl Cytoxan (cyclophosphamide) -

Chlorambucil
Chlorambucil DRUG NAME: Chlorambucil SYNONYM(S): Chlorambucilum,1 Chloraminophene,2 Chlorbutinum,1 CB-1348,1, NSC-30881 COMMON TRADE NAME(S): LEUKERAN® CLASSIFICATION: alkylating agent Special pediatric considerations are noted when applicable, otherwise adult provisions apply. MECHANISM OF ACTION: Chlorambucil is a derivative of nitrogen mustard and acts as a cell cycle phase-nonspecific bifunctional alkylating agent.3,4 Alkylation takes place through the formation of a highly reactive ethylenimonium radical.3 This radical likely forms a cross-linkage between two strands of DNA, interfering with DNA, RNA and protein synthesis.3,5 Chlorambucil also demonstrates immunosuppressive activity principally due to its suppression of lymphocytes.5 PHARMACOKINETICS: Oral Absorption 70-80%,2 rapidly and completely absorbed,5 bioavailability reduced by 10-20% with food6 Distribution to liver, ascitic fluid, fat, crosses the placenta7 cross blood brain barrier? no information found volume of distribution6 0.14-0.24 L/kg plasma protein binding 99% Metabolism primarily hepatic active metabolite(s) phenylacetic acid mustard inactive metabolite(s) monohydroxy and dihydroxy derivatives Excretion urine low urinary excretion, as almost completely metabolized feces no information found terminal half life3,5,6 1.5 h; 1.8-2.5 h phenylacetic acid mustard clearance8 0.16 + 0.04 L/hr/kg Adapted from standard reference3 unless specified otherwise. USES: Primary uses: Other uses: *Chronic lymphocytic leukemia Gestational trophoblastic tumour6 *Hodgkin’s lymphoma -

Febuxostat for Management of Tumor Lysis Syndrome Including Its Effects
ANTICANCER RESEARCH 34: 7287-7296 (2014) Febuxostat for Management of Tumor Lysis Syndrome Including its Effects on Levels of Purine Metabolites in Patients with Hematological Malignancies - A Single Institution’s, Pharmacokinetic and Pilot Prospective Study - MIHOKO TAKAI, TAKAHIRO YAMAUCHI, MIYUKI OOKURA, YASUFUMI MATSUDA, KATSUNORI TAI, SHINJI KISHI, AKIRA YOSHIDA, HIROMICHI IWASAKI, TORU NAKAMURA and TAKANORI UEDA Department of Hematology and Oncology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan Abstract. Background/Aim: Tumor lysis syndrome (TLS) is elevated along with the decrease in S-UA. Xanthine level was a life-threatening oncological emergency, and control of elevated higher compared to hypoxanthine level and reached serum uric acid level (S-UA) is most important. In this the level reported to cause xanthine nephropathy, but no single-institution, short-term and pilot prospective study, the advance of renal impairment was observed. Serum febuxostat efficacy of a new xanthine oxidase inhibitor, febuxostat, as concentrations at 2 h after administration were 891.8±285.0 an alternative to conventional allopurinol, including its ng/ml (mean±SE) for the 40-mg dose and 770.6±242.7 ng/ml effects on hypoxanthine and xanthine, was evaluated in 10 for the 60-mg dose (p=0.80, unpaired t-test), showing no consecutive patients with hematological malignancies at accumulation in patients with renal impairment. No intermediate risk for TLS. Patients and Methods: Febuxostat febuxostat-related adverse reactions were noted. No patients at 40 mg (n=7) or 60 mg (n=3) daily was administered experienced progressive TLS. Conclusion: Febuxostat is according to renal function, and induction chemotherapy was promising for the management of TLS of an intermediate-risk started within 24 h. -

Treating Non-Hodgkin Lymphoma If You’Ve Been Diagnosed with Non-Hodgkin Lymphoma, Your Treatment Team Will Discuss Your Options with You
cancer.org | 1.800.227.2345 Treating Non-Hodgkin Lymphoma If you’ve been diagnosed with non-Hodgkin lymphoma, your treatment team will discuss your options with you. It’s important to weigh the benefits of each treatment option against the possible risks and side effects. How is non-Hodgkin lymphoma treated? Depending on the type and stage (extent) of the lymphoma and other factors, treatment options for people with NHL might include: ● Chemotherapy for Non-Hodgkin Lymphoma ● Immunotherapy for Non-Hodgkin Lymphoma ● Targeted Drug Therapy for Non-Hodgkin Lymphoma ● Radiation Therapy for Non-Hodgkin Lymphoma ● High-Dose Chemotherapy and Stem Cell Transplant for Non-Hodgkin Lymphoma ● Surgery for Non-Hodgkin Lymphoma Common treatment approaches Treatment approaches for NHL depend on the type of cancer, how advanced it is, as well as your health and other factors. Another important part of treatment for many people is palliative or supportive care. This can help prevent or treat problems such as infections, low blood cell counts, or some symptoms caused by the lymphoma. ● Treating B-Cell Non-Hodgkin Lymphoma ● Treating T-Cell Non-Hodgkin Lymphoma ● Treating HIV-Associated Lymphoma 1 ____________________________________________________________________________________American Cancer Society cancer.org | 1.800.227.2345 ● Palliative and Supportive Care for Non-Hodgkin Lymphoma Who treats non-Hodgkin lymphoma? Based on your treatment options, you may have different types of doctors on your treatment team. These doctors could include: ● A medical oncologist or hematologist: a doctor who treats lymphoma with chemotherapy, immunotherapy, and targeted therapy. ● A radiation oncologist: a doctor who treats cancer with radiation therapy. ● A bone marrow transplant doctor: a doctor who specializes in treating cancer or other diseases with bone marrow or stem cell transplants.