Detection and Characterization of Low and High Genome Coverage Regions Using an Efficient Running Median and a Double Threshold Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
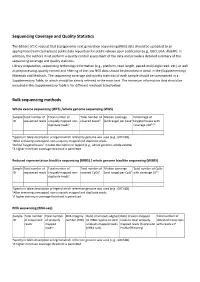
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Transcriptome Analysis Using Next-Generation Sequencing
Available online at www.sciencedirect.com Transcriptome analysis using next-generation sequencing Kai-Oliver Mutz, Alexandra Heilkenbrinker, Maren Lo¨ nne, Johanna-Gabriela Walter and Frank Stahl Up to date research in biology, biotechnology, and medicine more than 15 years and millions of dollars, today it is requires fast genome and transcriptome analysis technologies possible to sequence it in about eight days for approxi- for the investigation of cellular state, physiology, and activity. mately $100 000 [4]. The important technological devel- Here, microarray technology and next generation sequencing opments, summarized under the term next-generation of transcripts (RNA-Seq) are state of the art. Since microarray sequencing, are based on the sequencing-by-synthesis technology is limited towards the amount of RNA, the (SBS) technology called pyrosequencing. The transcrip- quantification of transcript levels and the sequence tomics variant of pyrosequencing technology is called information, RNA-Seq provides nearly unlimited possibilities in short-read massively parallel sequencing or RNA-Seq modern bioanalysis. This chapter presents a detailed [5]. In recent years, RNA-Seq is rapidly emerging as description of next-generation sequencing (NGS), describes the major quantitative transcriptome profiling system the impact of this technology on transcriptome analysis and [6,7]. Different companies developed different sequen- explains its possibilities to explore the modern RNA world. cing platforms all based on variations of pyrosequencing. Address Next-generation sequencing Leibniz Universita¨ t Hannover, Institute for Technical Chemistry, To achieve sequence information, various methods are Callinstrasse 5, 30167 Hannover, Germany well established today. The classical dideoxy method, Corresponding author: Stahl, Frank ([email protected]) invented by Friedrich Sanger in the 1970th uses an enzymatic reaction. -

An Introduction to Next-Generation Sequencing Technology
An introduction to Next-Generation Sequencing Technology www.illumina.com/technology/next-generation-sequencing.html For Research Use Only. Not for use in diagnostic procedures. Table of Contents Table of Contents 2 I. Welcome to Next-Generation Sequencing 3 a. The Evolution of Genomic Science 3 b. The Basics of NGS Chemistry 4 c. Advances in Sequencing Technology 5 Paired-End Sequencing 5 Tunable Coverage and Unlimited Dynamic Range 6 Advances in Library Preparation 6 Multiplexing 7 Flexible, Scalable Instrumentation 7 II. NGS Methods 8 a. Genomics 8 Whole-Genome Sequencing 8 Exome Sequencing 8 De novo Sequencing 9 Targeted Sequencing 9 b. Transcriptomics 11 Total RNA and mRNA Sequencing 11 Targeted RNA Sequencing 11 Small RNA and Noncoding RNASequencing 11 c. Epigenomics 12 Methylation Sequencing 12 ChIP Sequencing 12 Ribosome Profiling 12 III. Illumina DNA-to-Data NGS Solutions 13 a. The Illumina NGS Workflow 13 b. Integrated Data Analysis 13 IV. Glossary 14 V. References 15 For Research Use Only. Not for use in diagnostic procedures. I. Welcome to Next-Generation Sequencing a. The Evolution of Genomic Science DNA sequencing has come a long way since the days of two-dimensional chromatography in the 1970s. With the advent of the Sanger chain termination method1 in 1977, scientists gained the ability to sequence DNA in a reliable, reproducible manner. A decade later, Applied Biosystems introduced the first automated, capillary electrophoresis (CE)-based sequencing instruments,the AB370 in 1987 and the AB3730xl in 1998,instruments that became the primary workhorses for the NIH-led and Celera-led Human Genome Projects.2 While these “first-generation” instruments were considered high throughput for their time, the Genome Analyzer emerged in 2005 and took sequencing runs from 84 kilobase (kb) per run to 1 gigabase (Gb) per run.3 The short read, massively parallel sequencing technique was a fundamentally different approach that revolutionized sequencing capabilities and launched the “next generation” in genomic science. -

Information Theory of DNA Shotgun Sequencing Abolfazl S
IEEE TRANSACTIONS ON INFORMATION THEORY, VOL. 59, NO. 10, OCTOBER 2013 6273 Information Theory of DNA Shotgun Sequencing Abolfazl S. Motahari, Guy Bresler, Student Member, IEEE, and David N. C. Tse, Fellow, IEEE Abstract—DNA sequencing is the basic workhorse of modern day biology and medicine. Shotgun sequencing is the dominant technique used: many randomly located short fragments called reads are extracted from the DNA sequence, and these reads are assembled to reconstruct the original sequence. A basic question is: given a sequencing technology and the statistics of the DNA sequence, what is the minimum number of reads required for reliable reconstruction? This number provides a fundamental Fig. 1. Schematic for shotgun sequencing. limit to the performance of any assembly algorithm. For a simple statistical model of the DNA sequence and the read process, we show that the answer admits a critical phenomenon in the asymp- totic limit of long DNA sequences: if the read length is below Shotgun sequencing is the dominant method currently used a threshold, reconstruction is impossible no matter how many to sequence long strands of DNA, including entire genomes. reads are observed, and if the read length is above the threshold, The basic shotgun DNA sequencing setup is shown in Fig. 1. having enough reads to cover the DNA sequence is sufficient to Starting with a DNA molecule, the goal is to obtain the sequence reconstruct. The threshold is computed in terms of the Renyi entropy rate of the DNA sequence. We also study the impact of of nucleotides ( , , ,or ) comprising it. (For humans, the noise in the read process on the performance. -

1 G&T-Seq: Separation and Parallel Sequencing of the Genomes And
G&T-seq: Separation and parallel sequencing of the genomes and transcriptomes of single cells Iain C. Macaulay1, Wilfried Haerty2*, Parveen Kumar3*, Yang I. Li2†, Tim Xiaoming Hu2, Mabel J. Teng4, Mubeen Goolam5, Nathalie Saurat6, Paul Coupland7, Lesley M. Shirley7, Miriam Smith7, Niels Van der Aa3, Ruby Banerjee8, Peter D. Ellis7, Michael A. Quail7, Harold P. Swerdlow7‡, Magdalena Zernicka- Goetz5, Frederick J. Livesey6, Chris P. Ponting1,2^, Thierry Voet1,3^ * equal contribution ^ equal contribution Affiliations: 1Sanger Institute-EBI Single-Cell Genomics Centre, Wellcome Trust Sanger Institute, Hinxton, CB10 1SA, UK 2MRC Functional Genomics Unit, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, OX1 3QX, UK. 3Department of Human Genetics, University of Leuven, KU Leuven, Leuven, 3000 Belgium 4Cancer Genome Project, Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1SA, UK 5Department of Physiology, Development and Neuroscience, Downing Site, University of Cambridge, Cambridge, CB2 3DY, UK. 6Wellcome Trust/Cancer Research UK Gurdon Institute, University of Cambridge, Cambridge, CB2 1QN, UK 7Sequencing R&D, Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1SA, UK 8Cytogenetics core facility, Wellcome Trust Sanger Institute, Hinxton, Cambridge, CB10 1SA, UK 1 †Present address: Department of Genetics, Stanford University, Stanford, CA, 94305, USA ‡Present address: New York Genome Center, 101 Ave. of the Americas 7th Fl., New York, NY, 10013, USA 2 Abstract: The ability to simultaneously sequence the genome and transcriptome of the same single cell offers a powerful means to dissect functional genetic heterogeneity at the cellular level. Here we describe G&T-seq, a method for separating and sequencing genomic DNA and full-length mRNA from single cells. -
Comparison of Transcriptomics Technologies for Assessment of Staphylococcus Aureus Gene Expression
Comparison of transcriptomics technologies for assessment of Staphylococcus aureus gene expression Saturday, May 10 • P 0196 POSTERS D. Hernandez1, D.Baud 1, J. Schrenzel 1, P. François 1, A. Fischer 1, M. Girard 1, J.B. Veyrieras 2, C. Le Priol 2, G. Gervasi 2, F. Reynier 2 1Genomic Research Laboratory, Infectious Diseases Service, Geneva University Hospitals, Geneva, Switzerland, 2bioMérieux, Technology Research Department, Innovation and Systems Unit, Marcy l’Etoile, France. Objectives: Today, high-throughput shotgun sequencing of transcriptomes (RNA-seq) in prokaryotes seems to be an interesting alternative to well- established transcriptomics technologies such as microarray. While this later technology provides an analogical quantification of gene transcription (via the fluorescent intensity measuring the amount of hybridization between capture probes and their complementary cDNA fragments), RNA-seq methods make it possible to obtain a comprehensive digital quantification of transcribed regions (by counting the number of sequenced reads that map onto the corresponding genomic regions). Furthermore, contrary to existing digital technologies like the NanoString nCounter platform (and contrary to microarrays too), RNA-seq approaches do not require the prior design of probes and can then be used to simultaneously determine the transcriptomic profile of prokaryote strains at both known and unknown transcribed regions (Baume, et al., 2010). Nevertheless, the analytical performance of RNA-seq approaches in prokaryotes has not yet been investigated. Here, we compared two RNA-seq solutions (Illumina MiSeq and Ion Torrent PGM) with Agilent microarrays and the NanoString nCounter system on Staphylococcus aureus total RNA samples. Methods: We extracted four total RNA samples from the Staphylococcus aureus strain NCTC 8325. -
Exome Versus Transcriptome Sequencing in Identifying Coding Region Variants
THEMED ARTICLE S Genetic & Genomics Applications Review For reprint orders, please contact [email protected] Exome versus transcriptome sequencing in identifying coding region variants Expert Rev. Mol. Diagn. 12(3), 241–251 (2012) Chee-Seng Ku*1, The advent of next-generation sequencing technologies has revolutionized the study of genetic Mengchu Wu1, variation in the human genome. Whole-genome sequencing currently represents the most David N Cooper2, comprehensive strategy for variant detection genome-wide but is costly for large sample sizes, Nasheen Naidoo3, and variants detected in noncoding regions remain largely uninterpretable. By contrast, whole- 4 exome sequencing has been widely applied in the identification of germline mutations underlying Yudi Pawitan , Mendelian disorders, somatic mutations in various cancers and de novo mutations in 5 Brendan Pang , neurodevelopmental disorders. Since whole-exome sequencing focuses upon the entire set of Barry Iacopetta6 and exons in the genome (the exome), it requires additional exome-enrichment steps compared Richie Soong1 with whole-genome sequencing. Although the availability of multiple commercial exome-enrichment 1Cancer Science Institute of Singapore kits has made whole-exome sequencing technically feasible, it has also added to the overall (CSI Singapore), #12-01, MD6, Centre cost. This has led to the emergence of transcriptome (or RNA) sequencing as a potential for Translational Medicine, NUS Yong alternative approach to variant detection within protein coding regions, since the transcriptome Loo Lin School of Medicine, National of a given tissue represents a quasi-complete set of transcribed genes (mRNAs) and other University of Singapore, 14 Medical Drive, 117599, Singapore noncoding RNAs. A further advantage of this approach is that it bypasses the need for exome 2Institute of Medical Genetics, School enrichment. -

Cfdna Sequencing: Technological Approaches and Bioinformatic Issues
pharmaceuticals Review cfDNA Sequencing: Technological Approaches and Bioinformatic Issues Elodie Bohers * , Pierre-Julien Viailly and Fabrice Jardin INSERM U1245, Henri Becquerel Center, IRIB, Normandy University, 76000 Rouen, France; [email protected] (P.-J.V.); [email protected] (F.J.) * Correspondence: [email protected] Abstract: In the era of precision medicine, it is crucial to identify molecular alterations that will guide the therapeutic management of patients. In this context, circulating tumoral DNA (ctDNA) released by the tumor in body fluids, like blood, and carrying its molecular characteristics is becoming a powerful biomarker for non-invasive detection and monitoring of cancer. Major recent technological advances, especially in terms of sequencing, have made possible its analysis, the challenge still being its reliable early detection. Different parameters, from the pre-analytical phase to the choice of sequencing technology and bioinformatic tools can influence the sensitivity of ctDNA detection. Keywords: cell-free DNA; circulating tumoral DNA; sequencing technologies; bioinformatics 1. Introduction Cell free circulating DNA (cfDNA) refers to DNA fragments present outside of cells in body fluids such as plasma, urine, and cerebrospinal fluid (CSF). CfDNA was first Citation: Bohers, E.; Viailly, P.-J.; identified in 1948 from plasma of healthy individuals [1]. Afterward, studies showed that Jardin, F. cfDNA Sequencing: the quantity of this cfDNA in the blood was increased under pathological conditions such Technological Approaches and as auto-immune diseases [2] but also cancers [3]. In 1989, Philippe Anker and Maurice Bioinformatic Issues. Pharmaceuticals Stroun, from the University of Geneva, demonstrated that this cfDNA from cancer patients 2021, 14, 596. -

Benchmarking Full-Length Transcript Single Cell Mrna Sequencing Protocols
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.29.225201; this version posted August 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Benchmarking full-length transcript single cell mRNA sequencing protocols Victoria Probst1, Felix Pacheco1, Finn Cilius Nielsen1, Frederik Otzen Bagger1,* 1Genomic Medicine, Rigshospitalet, University of Copenhagen, Denmark *Correspondence to: Frederik Otzen Bagger ([email protected]) bioRxiv preprint doi: https://doi.org/10.1101/2020.07.29.225201; this version posted August 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. - 1 - Abstract Single cell mRNA sequencing technologies have transformed our understanding of cellular heterogeneity and identity. For sensitive discovery or clinical marker estimation where high transcript capture per cell is needed only plate-based techniques currently offer sufficient resolution. Here, we present a performance evaluation of three different plate-based scRNA-seq protocols. Our evaluation is aimed towards applications requiring high gene detection sensitivity, reproducibility between samples, and minimum hands-on time, as is required, for example, in clinical use. We included two commercial kits, NEBNextÒ Single Cell/ Low Input RNA Library Prep Kit (NEBÒ), SMART-seqÒ HT kit (TakaraÒ), and the non-commercial protocol Genome & Transcriptome sequencing (G&T). -

Whole Exome and Whole Genome Sequencing
UnitedHealthcare® Commercial Medical Policy Whole Exome and Whole Genome Sequencing Policy Number: 2021T0589I Effective Date: January 1, 2021 Instructions for Use Table of Contents Page Related Commercial Policies Coverage Rationale ........................................................................... 1 • Chromosome Microarray Testing (Non-Oncology Documentation Requirements......................................................... 2 Conditions) Definitions ........................................................................................... 2 • Molecular Oncology Testing for Cancer Diagnosis, Applicable Codes .............................................................................. 3 Prognosis, and Treatment Decisions Description of Services ..................................................................... 4 • Preimplantation Genetic Testing Clinical Evidence ............................................................................... 4 U.S. Food and Drug Administration ..............................................22 Community Plan Policy References .......................................................................................22 • Whole Exome and Whole Genome Sequencing Policy History/Revision Information..............................................26 Medicare Advantage Coverage Summaries Instructions for Use .........................................................................27 • Genetic Testing • Laboratory Tests and Services Coverage Rationale Whole Exome Sequencing (WES) Whole Exome Sequencing -

Guidelines for the Experimental Design of Single-Cell RNA Sequencing Studies
REVIEW ARTICLE https://doi.org/10.1038/s41596-018-0073-y Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies Atefeh Lafzi1,5, Catia Moutinho1,5, Simone Picelli2,4, Holger Heyn 1,3* Single-cell RNA sequencing is at the forefront of high-resolution phenotyping experiments for complex samples. Although this methodology requires specialized equipment and expertise, it is now widely applied in research. However, it is challenging to create broadly applicable experimental designs because each experiment requires the user to make informed decisions about sample preparation, RNA sequencing and data analysis. To facilitate this decision-making process, in this tutorial we summarize current methodological and analytical options, and discuss their suitability for a range of research scenarios. Specifically, we provide information about best practices for the separation of individual cells and provide an overview of current single-cell capture methods at different cellular resolutions and scales. Methods for the preparation of RNA sequencing libraries vary profoundly across applications, and we discuss features important for an informed selection process. An erroneous or biased analysis can lead to misinterpretations or obscure biologically important information. We provide a guide to the major data processing steps and options for meaningful data interpretation. These guidelines will serve as a reference to support users in building a single-cell experimental framework —from sample preparation to data interpretation—that is tailored to the underlying research context. 1234567890():,; 1234567890():,; ingle-cell transcriptomics studies have markedly a result, single-cell research has become one of the fastest- fi improved our understanding of the complexity of tissues, growing elds in life science, producing fascinating new S 1 fi insights into tissue composition and dynamic biological pro- organs and organisms . -

Transcriptomics Technologies
TOPIC PAGE Transcriptomics technologies Rohan Lowe1, Neil Shirley2, Mark Bleackley1, Stephen Dolan3, Thomas Shafee1* 1 La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Australia, 2 ARC Centre of Excellence in Plant Cell Walls, University of Adelaide, Adelaide, Australia, 3 Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom * [email protected] Abstract Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst noncoding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. The first attempts to study the whole transcriptome began in the early 1990s, and techno- logical advances since the late 1990s have made transcriptomics a widespread discipline. a1111111111 Transcriptomics has been defined by repeated technological innovations that transform the a1111111111 field. There are two key contemporary techniques in the field: microarrays, which quantify a a1111111111 a1111111111 set of predetermined sequences, and RNA sequencing (RNA-Seq), which uses high- a1111111111 throughput sequencing to capture all sequences. Measuring the expression of an organism's genes in different tissues, conditions, or time points gives information on how genes are regulated and reveals details of an organism's biology. It can also help to infer the functions of previously unannotated genes. Transcrip- OPEN ACCESS tomic analysis has enabled the study of how gene expression changes in different organ- isms and has been instrumental in the understanding of human disease.