Exome Versus Transcriptome Sequencing in Identifying Coding Region Variants
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Whole Exome and Whole Genome Sequencing – Oxford Clinical Policy
UnitedHealthcare® Oxford Clinical Policy Whole Exome and Whole Genome Sequencing Policy Number: LABORATORY 024.11 T2 Effective Date: October 1, 2021 Instructions for Use Table of Contents Page Related Policies Coverage Rationale ....................................................................... 1 Chromosome Microarray Testing (Non-Oncology Documentation Requirements ...................................................... 2 Conditions) Definitions ...................................................................................... 2 Molecular Oncology Testing for Cancer Diagnosis, Prior Authorization Requirements ................................................ 3 Prognosis, and Treatment Decisions Applicable Codes .......................................................................... 3 • Preimplantation Genetic Testing Description of Services ................................................................. 4 Clinical Evidence ........................................................................... 4 U.S. Food and Drug Administration ........................................... 22 References ................................................................................... 22 Policy History/Revision Information ........................................... 26 Instructions for Use ..................................................................... 27 Coverage Rationale Whole Exome Sequencing (WES) Whole Exome Sequencing (WES) is proven and Medically Necessary for the following: • Diagnosing or evaluating a genetic disorder -

Whole Exome Sequencing Faqs
WHOLE EXOME SEQUENCING (WES) Whole Exome Sequencing (WES) is generally ordered when a patient’s medical history and physical exam strongly suggest that there is an underlying genetic etiology. In some cases, the patient may have had an extensive evaluation consisting of multiple genetic tests, without identifying an etiology. In other cases, a physician may opt to order one of the Whole Exome Sequencing tests early in the patient’s evaluation in an effort to expedite a possible diagnosis and reduce costs incurred by multiple tests. Whole Exome Sequencing is a highly complex test that is newly developed for the identification of changes in a patient’s DNA that are causative or related to their medical concerns. In contrast to current sequencing tests that analyze one gene or small groups of related genes at a time, Whole Exome Sequencing analyzes the exons or coding regions of thousands of genes simultaneously using next-generation sequencing techniques. The exome refers to the portion of the human genome that contains functionally important sequences of DNA that direct the body to make proteins essential for the body to function properly. These regions of DNA are referred to as exons. There are approximately 180,000 exons in the human genome which represents about 3% of the genome. These 180,000 exons are arranged in about 22,000 genes. It is known that many of the errors that occur in DNA sequences that then lead to genetic disorders are located in the exons. Therefore, sequencing of the exome is thought to be an efficient method of analyzing a patient’s DNA to discover the genetic cause of diseases or disabilities. -
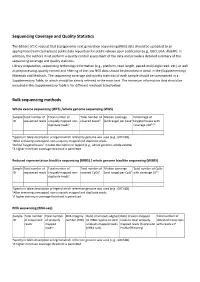
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Microbes and Metagenomics in Human Health an Overview of Recent Publications Featuring Illumina® Technology TABLE of CONTENTS
Microbes and Metagenomics in Human Health An overview of recent publications featuring Illumina® technology TABLE OF CONTENTS 4 Introduction 5 Human Microbiome Gut Microbiome Gut Microbiome and Disease Inflammatory Bowel Disease (IBD) Metabolic Diseases: Diabetes and Obesity Obesity Oral Microbiome Other Human Biomes 25 Viromes and Human Health Viral Populations Viral Zoonotic Reservoirs DNA Viruses RNA Viruses Human Viral Pathogens Phages Virus Vaccine Development 44 Microbial Pathogenesis Important Microorganisms in Human Health Antimicrobial Resistance Bacterial Vaccines 54 Microbial Populations Amplicon Sequencing 16S: Ribosomal RNA Metagenome Sequencing: Whole-Genome Shotgun Metagenomics Eukaryotes Single-Cell Sequencing (SCS) Plasmidome Transcriptome Sequencing 63 Glossary of Terms 64 Bibliography This document highlights recent publications that demonstrate the use of Illumina technologies in immunology research. To learn more about the platforms and assays cited, visit www.illumina.com. An overview of recent publications featuring Illumina technology 3 INTRODUCTION The study of microbes in human health traditionally focused on identifying and 1. Roca I., Akova M., Baquero F., Carlet J., treating pathogens in patients, usually with antibiotics. The rise of antibiotic Cavaleri M., et al. (2015) The global threat of resistance and an increasingly dense—and mobile—global population is forcing a antimicrobial resistance: science for interven- tion. New Microbes New Infect 6: 22-29 1, 2, 3 change in that paradigm. Improvements in high-throughput sequencing, also 2. Shallcross L. J., Howard S. J., Fowler T. and called next-generation sequencing (NGS), allow a holistic approach to managing Davies S. C. (2015) Tackling the threat of anti- microbial resistance: from policy to sustainable microbes in human health. -

Bioinformatics Tools for RNA-Seq Gene and Isoform Quantification
on: Sequ ati en er c n in e g G & t x A Journal of e p Zhang, et al., Next Generat Sequenc & Applic p N l f i c o 2016, 3:3 a l t a i o n r ISSN: 2469-9853n u s DOI: 10.4172/2469-9853.1000140 o Next Generation Sequencing & Applications J Review Article Open Access Bioinformatics Tools for RNA-seq Gene and Isoform Quantification Chi Zhang1, Baohong Zhang1, Michael S Vincent2 and Shanrong Zhao1* 1Early Clinical Development, Pfizer Worldwide R&D, Cambridge, MA, USA 2Inflammation and Immunology RU, Pfizer Worldwide R&D, Cambridge, MA, USA *Corresponding author: Shanrong Zhao, Early Clinical Development, Pfizer Worldwide R&D, Cambridge, MA, 02139, USA, Tel: + 1-212-733-2323; E-mail: [email protected] Rec date: Oct 27, 2016; Acc date: Dec 15, 2016; Pub date: Dec 17, 2016 Copyright: © 2016 Zhang C, et al. This is an open-access article distributed under the terms of the creative commons attribution license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract In recent years, RNA-seq has emerged as a powerful technology in estimation of gene or transcript expression. ‘Union-exon’ and transcript based approaches are widely used in gene quantification. The ‘Union-exon’ based approach is simple, but it does not distinguish between isoforms when multiple alternatively spliced transcripts are expressed from the same gene. Because a gene is expressed in one or more transcript isoforms, the transcript based approach is more biologically meaningful than the ‘union exon’-based approach. -

Next-Gen Sequencing Identifies Non-Coding Variation Disrupting
OPEN Molecular Psychiatry (2018) 23, 1375–1384 www.nature.com/mp ORIGINAL ARTICLE Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders P Devanna1, XS Chen2,JHo1,2, D Gajewski1, SD Smith3, A Gialluisi2,4, C Francks2,5, SE Fisher2,5, DF Newbury6,7 and SC Vernes1,5 Understanding the genetic factors underlying neurodevelopmental and neuropsychiatric disorders is a major challenge given their prevalence and potential severity for quality of life. While large-scale genomic screens have made major advances in this area, for many disorders the genetic underpinnings are complex and poorly understood. To date the field has focused predominantly on protein coding variation, but given the importance of tightly controlled gene expression for normal brain development and disorder, variation that affects non-coding regulatory regions of the genome is likely to play an important role in these phenotypes. Herein we show the importance of 3 prime untranslated region (3'UTR) non-coding regulatory variants across neurodevelopmental and neuropsychiatric disorders. We devised a pipeline for identifying and functionally validating putatively pathogenic variants from next generation sequencing (NGS) data. We applied this pipeline to a cohort of children with severe specific language impairment (SLI) and identified a functional, SLI-associated variant affecting gene regulation in cells and post-mortem human brain. This variant and the affected gene (ARHGEF39) represent new putative risk factors for SLI. Furthermore, we identified 3′UTR regulatory variants across autism, schizophrenia and bipolar disorder NGS cohorts demonstrating their impact on neurodevelopmental and neuropsychiatric disorders. Our findings show the importance of investigating non-coding regulatory variants when determining risk factors contributing to neurodevelopmental and neuropsychiatric disorders. -

An Efficient and Scalable Analysis Framework for Variant Extraction and Refinement from Population-Scale DNA Sequence Data
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Method An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data Goo Jun,1,2 Mary Kate Wing,2 Gonçalo R. Abecasis,2 and Hyun Min Kang2 1Human Genetics Center, School of Public Health, The University of Texas Health Science Center at Houston, Houston, Texas 77030, USA; 2Center for Statistical Genetics and Department of Biostatistics, University of Michigan School of Public Health, Ann Arbor, Michigan 48109, USA The analysis of next-generation sequencing data is computationally and statistically challenging because of the massive vol- ume of data and imperfect data quality. We present GotCloud, a pipeline for efficiently detecting and genotyping high- quality variants from large-scale sequencing data. GotCloud automates sequence alignment, sample-level quality control, variant calling, filtering of likely artifacts using machine-learning techniques, and genotype refinement using haplotype in- formation. The pipeline can process thousands of samples in parallel and requires less computational resources than current alternatives. Experiments with whole-genome and exome-targeted sequence data generated by the 1000 Genomes Project show that the pipeline provides effective filtering against false positive variants and high power to detect true variants. Our pipeline has already contributed to variant detection and genotyping in several large-scale sequencing projects, including the 1000 Genomes Project and the NHLBI Exome Sequencing Project. We hope it will now prove useful to many medical sequencing studies. [Supplemental material is available for this article.] The cost of human genome sequencing has declined rapidly, pow- There is a pressing need for software pipelines that support ered by advances in massively parallel sequencing technologies. -

Whole Exome and Whole Genome Sequencing
UnitedHealthcare® Community Plan Medical Policy Whole Exome and Whole Genome Sequencing Policy Number: CS150.J Effective Date: October 1, 2021 Instructions for Use Table of Contents Page Related Community Plan Policies Application ..................................................................................... 1 • Chromosome Microarray Testing (Non-Oncology Coverage Rationale ....................................................................... 1 Conditions) Definitions ...................................................................................... 2 • Molecular Oncology Testing for Cancer Diagnosis, Applicable Codes .......................................................................... 3 Prognosis, and Treatment Decisions Description of Services ................................................................. 4 • Preimplantation Genetic Testing Clinical Evidence ........................................................................... 4 U.S. Food and Drug Administration ........................................... 22 Commercial Policy References ................................................................................... 22 • Whole Exome and Whole Genome Sequencing Policy History/Revision Information ........................................... 26 Medicare Advantage Coverage Summaries Instructions for Use ..................................................................... 26 • Genetic Testing • Laboratory Tests and Services Application This Medical Policy does not apply to the states listed below; refer to -

Clinical Exome Sequencing Tip Sheet – Medicare Item Numbers 73358/73359
Clinical exome sequencing Tip sheet – Medicare item numbers 73358/73359 Glossary Chromosome microarray (CMA or molecular Monogenic conditions (as opposed karyotype): CMA has a Medicare item number to polygenic or multifactorial conditions) are for patients presenting with intellectual caused by variants in a single gene. Variants disability, developmental delay, autism, or at may be inherited (dominant or recessive least two congenital anomalies. CMA is the fashion), or may occur spontaneously (de recommended first line test in these cases as novo) showing no family history. it can exclude a chromosome cause of disease which is unlikely to be detected by Whole exome sequence – sequencing only exome. the protein coding genes (exons). The exome is ~2% of the genome and contains ~85% of Gene panel is a set of genes that are known to disease-causing gene variants. be associated with a phenotype or disorder. They help narrow down the search Whole genome sequence – sequencing the for variants of interest to genes with evidence entire genome (all genes, including coding linking them to particular phenotypes and noncoding regions) Human phenotype ontology (HPO) terms Singleton – Analysis of the child only. describe a phenotypic abnormality using a Trio – analysis of the child and both biological standard nomenclature. Ideally, all clinicians parents. and scientists are using the same terms. Variant - A change in the DNA code that Mendeliome refers to the ~5,000 genes (out of differs from a reference genome. about 20,000 protein coding genes) that are known to be associated with monogenic disease. As variants in new genes are identified with evidence linking them with human disease, they are added to the Mendeliome. -

The Chromosome-Centric Human Proteome Project for Cataloging Proteins Encoded in the Genome
CORRESPONDENCE The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome To the Editor: utility for biological and disease studies. Table 1 Features of salient genes on The Chromosome-Centric Human With development of new tools for in- chromosomes 13 and 17 Proteome Project (C-HPP) aims to define depth characterization of the transcriptome Genea AST nsSNPs the full set of proteins encoded in each and proteome, the HPP is well positioned Chromosome 13 chromosome through development of a to have a strategic role in addressing the BRCA2 3 54 standardized approach for analyzing the complexity of human phenotypes. With this RB1 2 3 massive proteomic data sets currently being in mind, the HUPO has organized national IRS2 1 3 generated from dedicated efforts of national chromosome teams that will collaborate and international teams. The initial goal with well-established laboratories building Chromosome 17 of the C-HPP is to identify at least one complementary proteotypic peptides, BRCA1 24 24 representative protein encoded by each of antibodies and informatics resources. ERBB2 6 13 the approximately 20,300 human genes1,2. An important C-HPP goal is to encourage TP53 14 5 aEnsembl protein and AST information can be found at The proteins will be characterized for tissue capture and open sharing of proteomic http://www.ensembl.org/Homo_sapiens/. localization and major isoforms, including data sets from diverse samples to enhance AST, alternative splicing transcript; nsSNP, nonsyno- mous single-nucleotide polyphorphism assembled from post-translational modifications (PTMs), a gene- and chromosome-centric display data from the 1000 Genomes Projects. -

Downloaded As a CSV Dump file
cells Article Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis Nicolas Buisine 1,† , Alexis Grimaldi 1,†, Vincent Jonchere 1,† , Muriel Rigolet 1, Corinne Blugeon 2 , Juliette Hamroune 2 and Laurent Marc Sachs 1,* 1 UMR7221 Molecular Physiology and Adaption, CNRS, Museum National d’Histoire Naturelle, 57 Rue Cuvier, CEDEX 05, 75231 Paris, France; [email protected] (N.B.); [email protected] (A.G.); [email protected] (V.J.); [email protected] (M.R.) 2 Genomics Core Facility, Département de Biologie, Institut de Biologie de l’ENS (IBENS), École Normale Supérieure, CNRS, INSERM, Université PSL, 75005 Paris, France; [email protected] (C.B.); [email protected] (J.H.) * Correspondence: [email protected] † Co-first authors, alphabetic order. Abstract: Background: Most work in endocrinology focus on the action of a single hormone, and very little on the cross-talks between two hormones. Here we characterize the nature of interactions between thyroid hormone and glucocorticoid signaling during Xenopus tropicalis metamorphosis. Methods: We used functional genomics to derive genome wide profiles of methylated DNA and measured changes of gene expression after hormonal treatments of a highly responsive tissue, tailfin. Clustering classified the data into four types of biological responses, and biological networks were Citation: Buisine, N.; Grimaldi, A.; modeled by system biology. Results: We found that gene expression is mostly regulated by either Jonchere, V.; Rigolet, M.; Blugeon, C.; T or CORT, or their additive effect when they both regulate the same genes. A small but non- Hamroune, J.; Sachs, L.M. -

The Genomics Era: the Future of Genetics in Medicine - Glossary
The Genomics Era: the Future of Genetics in Medicine - Glossary The glossary below provides a list of key terms used throughout the course. You do not need to read them all now; we’ll be linking back to the main glossary step wherever these terms appear, so you may refer back to this list if you are unsure of the terminology being used. Term Definition The process of matching reads back to their original Alignment position in the reference genome. An allele is one of a number of alternative forms of the same gene or genetic locus. We inherit one copy Allele of our genetic code from our mother and one copy of our genetic code from our father. Each copy is known as an allele. Microarray based genomic comparative hybridisation. This is a technique used to detect chromosome imbalances by comparing patient and control DNA and comparing differences between the two sets. It is Array CGH a useful technique for detecting small chromosome deletions and duplications which would not have been detected with more traditional karyotyping techniques. A unit of DNA. There are four bases which form the Base cross links (or rungs) of the DNA double helix: adenine (A), thymine (T), guanine (G) and cytosine (C). Capture see Target enrichment. The process by which a cell becomes specialized in Cell differentiation order to perform a specific function. Centromere The point at which the sister chromatids are joined. #1 FutureLearn A structure located in the nucleus all living cells, comprised of DNA bound around proteins called histones. The normal number of chromosomes in each Chromosome human cell nucleus is 46 and is composed of 22 pairs of autosomes and a pair of sex chromosomes which determine gender: males have an X and a Y chromosome whilst females have two X chromosomes.