Single-Cell Multiomics: Technologies and Data Analysis Methods Jeongwoo Lee1,Doyounghyeon1 and Daehee Hwang1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
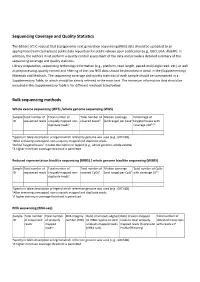
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine
biomolecules Review Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine Ryuji Hamamoto 1,2,*, Masaaki Komatsu 1,2, Ken Takasawa 1,2 , Ken Asada 1,2 and Syuzo Kaneko 1 1 Division of Molecular Modification and Cancer Biology, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan; [email protected] (M.K.); [email protected] (K.T.); [email protected] (K.A.); [email protected] (S.K.) 2 Cancer Translational Research Team, RIKEN Center for Advanced Intelligence Project, 1-4-1 Nihonbashi, Chuo-ku, Tokyo 103-0027, Japan * Correspondence: [email protected]; Tel.: +81-3-3547-5271 Received: 1 December 2019; Accepted: 27 December 2019; Published: 30 December 2019 Abstract: To clarify the mechanisms of diseases, such as cancer, studies analyzing genetic mutations have been actively conducted for a long time, and a large number of achievements have already been reported. Indeed, genomic medicine is considered the core discipline of precision medicine, and currently, the clinical application of cutting-edge genomic medicine aimed at improving the prevention, diagnosis and treatment of a wide range of diseases is promoted. However, although the Human Genome Project was completed in 2003 and large-scale genetic analyses have since been accomplished worldwide with the development of next-generation sequencing (NGS), explaining the mechanism of disease onset only using genetic variation has been recognized as difficult. Meanwhile, the importance of epigenetics, which describes inheritance by mechanisms other than the genomic DNA sequence, has recently attracted attention, and, in particular, many studies have reported the involvement of epigenetic deregulation in human cancer. -

CUT&Runtools: a Flexible Pipeline for CUT&RUN Processing and Footprint Analysis
Zhu et al. Genome Biology (2019) 20:192 https://doi.org/10.1186/s13059-019-1802-4 SOFTWARE Open Access CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis Qian Zhu1, Nan Liu2, Stuart H. Orkin2,3* and Guo-Cheng Yuan1* Abstract We introduce CUT&RUNTools as a flexible, general pipeline for facilitating the identification of chromatin-associated protein binding and genomic footprinting analysis from antibody-targeted CUT&RUN primary cleavage data. CUT&RUNTools extracts endonuclease cut site information from sequences of short-read fragments and produces single-locus binding estimates, aggregate motif footprints, and informative visualizations to support the high-resolution mapping capability of CUT&RUN. CUT&RUNTools is available at https://bitbucket.org/qzhudfci/cutruntools/. Introduction Results Mapping the occupancy of DNA-associated proteins, in- Overview cluding transcription factors (TFs) and histones, is central CUT&RUNTools takes paired-end sequencing read to determining cellular regulatory circuits. Conventional FASTQ files as the input and performs a set of analytical ChIP sequencing (ChIP-seq) relies on the cross-linking of steps: trimming of adapter sequences, alignment to the target proteins to DNA and physical fragmentation of reference genome, peak calling, estimation of cut matrix chromatin [1]. In practice, epitope masking and insolubil- at single-nucleotide resolution, de novo motif searching, ity of protein complexes may interfere with the successful motif footprinting analysis, direct binding site identifica- use of conventional ChIP-seq for some chromatin- tion, and data visualization (Fig. 1b). The outputs of the associated proteins [2–4]. CUT&RUN is a recently de- pipeline (Fig. 1c) are (1) an aggregate footprint capturing scribed native endonuclease-based method based on the the characteristics of chromatin-associated protein bind- binding of an antibody to a chromatin-associated protein ing (Fig. -

Multiomics Data Collection, Visualization, and Utilization for Guiding Metabolic Engineering
Lawrence Berkeley National Laboratory Recent Work Title Multiomics Data Collection, Visualization, and Utilization for Guiding Metabolic Engineering. Permalink https://escholarship.org/uc/item/51g19549 Authors Roy, Somtirtha Radivojevic, Tijana Forrer, Mark et al. Publication Date 2021 DOI 10.3389/fbioe.2021.612893 Peer reviewed eScholarship.org Powered by the California Digital Library University of California METHODS published: 09 February 2021 doi: 10.3389/fbioe.2021.612893 Multiomics Data Collection, Visualization, and Utilization for Guiding Metabolic Engineering Somtirtha Roy 1,2†, Tijana Radivojevic 1,2,3†, Mark Forrer 2,3,4, Jose Manuel Marti 1,2,3, Vamshi Jonnalagadda 1,2, Tyler Backman 1,3, William Morrell 2,3,4, Hector Plahar 1,2, Joonhoon Kim 3,5, Nathan Hillson 1,2,3 and Hector Garcia Martin 1,2,3,6* 1 Lawrence Berkeley National Laboratory, Biological Systems and Engineering Division, Berkeley, CA, United States, 2 Department of Energy, Agile BioFoundry, Emeryville, CA, United States, 3 Joint BioEnergy Institute, Emeryville, CA, United States, 4 Sandia National Laboratories, Biomaterials and Biomanufacturing, Livermore, CA, United States, 5 Chemical and Biological Processes Development Group, Pacific Northwest National Laboratory, Richland, WA, United States, 6 BCAM, Basque Center for Applied Mathematics, Bilbao, Spain Edited by: Biology has changed radically in the past two decades, growing from a purely descriptive Eduard Kerkhoven, Chalmers University of science into also a design science. The availability of tools that enable the precise Technology, Sweden modification of cells, as well as the ability to collect large amounts of multimodal data, Reviewed by: open the possibility of sophisticated bioengineering to produce fuels, specialty and Mario Andrea Marchisio, commodity chemicals, materials, and other renewable bioproducts. -

Multiomics Modeling of the Immunome, Transcriptome, Microbiome, Proteome and Metabolome Adaptations During Human Pregnancy
University of the Pacific Scholarly Commons Dugoni School of Dentistry Faculty Articles Arthur A. Dugoni School of Dentistry 1-1-2019 Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy Mohammad Sajjad Ghaemi Stanford University School of Medicine Daniel B. DiGiulio Stanford University School of Medicine Kévin Contrepois Stanford University School of Medicine Benjamin Callahan Stanford University School of Medicine Thuy T.M. Ngo Stanford University Follow this and additional works at: https://scholarlycommons.pacific.edu/dugoni-facarticles See P nextart of page the forMedicine additional and authorsHealth Sciences Commons Recommended Citation Ghaemi, M. S., DiGiulio, D. B., Contrepois, K., Callahan, B., Ngo, T. T., Lee-Mcmullen, B., Lehallier, B., Robaczewska, A., McIlwain, D., Rosenberg-Hasson, Y., Wong, R. J., Quaintance, C., Culos, A., Stanley, N., Tanada, A., Tsai, A., Gaudilliere, D., Ganio, E., Han, X., Ando, K., McNeil, L., Tingle, M., Wise, P., Maric, I., Sirota, M., Wyss-Coray, T., Winn, V. D., Druzin, M. L., & Gibbs, R. S. (2019). Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy. Bioinformatics, 35(1), 95–103. DOI: 10.1093/bioinformatics/bty537 https://scholarlycommons.pacific.edu/dugoni-facarticles/719 This Article is brought to you for free and open access by the Arthur A. Dugoni School of Dentistry at Scholarly Commons. It has been accepted for inclusion in Dugoni School of Dentistry Faculty Articles by an authorized administrator of Scholarly Commons. For more information, please contact [email protected]. Authors Mohammad Sajjad Ghaemi, Daniel B. DiGiulio, Kévin Contrepois, Benjamin Callahan, Thuy T.M. -

Differential Chromatin Binding of the Lung Lineage Transcription Factor NKX2-1 Resolves Opposing Murine Alveolar Cell Fates in Vivo ✉ Danielle R
ARTICLE https://doi.org/10.1038/s41467-021-22817-6 OPEN Differential chromatin binding of the lung lineage transcription factor NKX2-1 resolves opposing murine alveolar cell fates in vivo ✉ Danielle R. Little1,2, Anne M. Lynch1,3, Yun Yan 2, Haruhiko Akiyama4, Shioko Kimura 5 & Jichao Chen 1 Differential transcription of identical DNA sequences leads to distinct tissue lineages and then multiple cell types within a lineage, an epigenetic process central to progenitor and stem 1234567890():,; cell biology. The associated genome-wide changes, especially in native tissues, remain insufficiently understood, and are hereby addressed in the mouse lung, where the same lineage transcription factor NKX2-1 promotes the diametrically opposed alveolar type 1 (AT1) and AT2 cell fates. Here, we report that the cell-type-specific function of NKX2-1 is attributed to its differential chromatin binding that is acquired or retained during development in coordination with partner transcriptional factors. Loss of YAP/TAZ redirects NKX2-1 from its AT1-specific to AT2-specific binding sites, leading to transcriptionally exaggerated AT2 cells when deleted in progenitors or AT1-to-AT2 conversion when deleted after fate commitment. Nkx2-1 mutant AT1 and AT2 cells gain distinct chromatin accessible sites, including those specific to the opposite fate while adopting a gastrointestinal fate, suggesting an epigenetic plasticity unexpected from transcriptional changes. Our genomic analysis of single or purified cells, coupled with precision genetics, provides an epigenetic basis for alveolar cell fate and potential, and introduces an experimental benchmark for deciphering the in vivo function of lineage transcription factors. 1 Department of Pulmonary Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, USA. -

Chip Sequencing
DNBseqTM SERVICE OVERVIEW ChIP Sequencing Service Description Project Workflow ChIP Sequencing is widely used to analyze protein interactions with DNA. We care for your samples from the start through It combines chromatin immunoprecipitation (ChIP) with massively parallel to the result reporting. Highly experienced DNA sequencing to identify binding sites of DNA-associated proteins, laboratory professionals follow strict quality and can be used to precisely map global binding sites for any protein of procedures to ensure high quality results. abundant information in comparison with array-based ChIP-chip. SAMPLE PREPARATION and customized bioinformatics services to suit your specific research needs. Sample QC Confidence: Great correlation with HiSeq data. Low input: As low as 5ng ChIP-ed DNA/sample for human sample. Comprehensive analysis: Correlation analysis between ChIP-Seq and RNA-seq. LIBRARY CONSTRUCTION Sequencing Service Specification The BGI ChIP-Seq Service will be performed with BGI’s DNBseq Library QC sequencing technology, featuring combinatorial probe-anchor synthesis (cPAS) and DNA Nanoballs (DNB) technology[1] for superior data quality. SEQUENCING • 50bp Single-end sequencing reads • Standard output 20 Million reads per sample Sequencing QC • Clean data and bioinformatics analysis are available in standard file formats • Available data storage and bioinformatics RAW DATA OUTPUT applications • Cloud-based data storage and delivery system Data QC Turn Around Time BIOINFORMATICS • Typical 25 working days from sample QC acceptance ANALYSIS to final data delivery • Expedited services are available, contact your local BGI specialist for details Delivery QC DNBseqTM ChIP-Seq Data Analysis In addition to clean data output, BGI oers a range of standard, advanced and customized bioinformatics pipelines for your ChIP-Seq analysis project, including the correlation analysis of dierential expression genes and peak-related genes. -

REVIEW Genome-Wide Identification of DNA–Protein Interactions Using Chromatin Immunoprecipitation Coupled with Flow Cell Seque
1 REVIEW Genome-wide identification of DNA–protein interactions using chromatin immunoprecipitation coupled with flow cell sequencing Brad G Hoffman and Steven J M Jones1 Department of Cancer Endocrinology, BC Cancer Research Center, 675 West 10th Avenue, Vancouver, BC, Canada V5Z 1L3 1Micheal Smith Genome Sciences Centre, BC Cancer Agency, Suite 100-570 West 7th Avenue, Vancouver, BC, Canada V5Z 4S6 (Correspondence should be addressed to S J M Jones; Email: [email protected]) Abstract The transcriptional networks underlying mammalian cell performed, the information generated by ChIP-seq is development and function are largely unknown. The recently expected to allow the development of a framework for described use of flow cell sequencing devices in combination networks describing the transcriptional regulation of cellular with chromatin immunoprecipitation (ChIP-seq) stands to development and function. However, to date, this technology revolutionize the identification of DNA–protein interactions. has been applied only to a small number of cell types, and even As such, ChIP-seq is rapidly becoming the method of choice fewer tissues, suggesting a huge potential for novel discovery for the genome-wide localization of histone modifications in this field. and transcription factor binding sites. As further studies are Journal of Endocrinology (2009) 201, 1–13 Introduction sheared chromatin. An antibody specific to the protein of interest is then added to the sonicated material and DNA The transcriptional networks driving mammalian cell fragments bound to the protein of interest isolated via development and function are only beginning to be immunoprecipitation. DNA fragments are then released by elucidated. In many tissues transcription factors critical to reversing the cross-links and the fragments purified. -

Detection and Characterization of Low and High Genome Coverage Regions Using an Efficient Running Median and a Double Threshold Approach
bioRxiv preprint doi: https://doi.org/10.1101/092478; this version posted December 8, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC 4.0 International license. Bioinformatics doi.10.1093/bioinformatics/xxxxxx Advance Access Publication Date: 2 April 2015 Applications Note Genome analysis Detection and characterization of low and high genome coverage regions using an efficient running median and a double threshold approach. Dimitri Desvillechabrol 1;∗, Christiane Bouchier 2, Sean Kennedy 1, and Thomas Cokelaer 3∗ 1Institut Pasteur – Pole Biomics 2Institut Pasteur – Genomic Platform – Pole Biomics 3Institut Pasteur – Bioinformatics and Biostatistics Hub – C3BI, USR 3756 IP CNRS – Paris, France ∗To whom correspondence should be addressed. Associate Editor: XXXXXXX Received on XXXXX; revised on XXXXX; accepted on XXXXX Abstract Motivation: Next Generation Sequencing (NGS) provides researchers with powerful tools to investigate both prokaryotic and eukaryotic genetics. An accurate assessment of reads mapped to a specific genome consists of inspecting the genome coverage as number of reads mapped to a specific genome location. Most current methods use the average of the genome coverage (sequencing depth) to summarize the overall coverage. This metric quickly assess the sequencing quality but ignores valuable biological information like the presence of repetitive regions or deleted genes. The detection of such information may be challenging due to a wide spectrum of heterogeneous coverage regions, a mixture of underlying models or the presence of a non-constant trend along the genome. -

A Machine Learning Approach to Predict Metabolic Pathway Dynamics from Time-Series Multiomics Data
www.nature.com/npjsba ARTICLE OPEN A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data Zak Costello1,2,3 and Hector Garcia Martin1,2,3,4 New synthetic biology capabilities hold the promise of dramatically improving our ability to engineer biological systems. However, a fundamental hurdle in realizing this potential is our inability to accurately predict biological behavior after modifying the corresponding genotype. Kinetic models have traditionally been used to predict pathway dynamics in bioengineered systems, but they take significant time to develop, and rely heavily on domain expertise. Here, we show that the combination of machine learning and abundant multiomics data (proteomics and metabolomics) can be used to effectively predict pathway dynamics in an automated fashion. The new method outperforms a classical kinetic model, and produces qualitative and quantitative predictions that can be used to productively guide bioengineering efforts. This method systematically leverages arbitrary amounts of new data to improve predictions, and does not assume any particular interactions, but rather implicitly chooses the most predictive ones. npj Systems Biology and Applications (2018)4:19 ; doi:10.1038/s41540-018-0054-3 INTRODUCTION Mathematical modeling provides a systematic manner to Biology has been transformed in the second half of the twentieth leverage these data to predict the behavior of engineered century from a descriptive science to a design science. This systems. Hence, increasingly, computational biology is focusing transformation has been produced by a combination of the on large-scale modeling of dynamical systems predicting pheno- 18,19 discovery of DNA as the repository of genetic information,1 and of type from genotype. -

GETTING STARTED with Chip-SEQ
GETTING STARTED WITH ChIP-SEQ INTRODUCTION TO ChIP SEQ IN THIS GUIDE WE WILL INTRODUCE Chromatin-immunoprecipitation (ChIP) followed by ChIP SEQUENCING AND OUTLINE KEY sequencing of the immuno-precipitated DNA is a powerful STEPS OF THE EXPERIMENTAL PROCESS tool for the investigation of Protein:DNA interactions. To INCLUDING: perform ChIP-seq, chromatin is isolated from cells or tissues and fragmented. Antibodies against chromatin associated • Experimental design proteins are used to enrich for specific chromatin fragments. • Controls for ChIP-seq experiments The DNA is recovered, sequenced and aligned to a reference • Reference genome alignment of ChIP-seq reads genome to determine specific protein binding loci. ChIP (mapping) studies have increased our knowledge of transcription factor • Background estimation biology, DNA methylation and histone modifications. • Peak finding • Quality control of ChIP-seq experiments ChIP-seq was first described in 2007 (1). ChIP sequencing • Differential binding analysis (and also microRNA sequencing) was one of the first • Motif analysis methods to make use of the power of massively parallel or next-generation sequencing (NGS) to significantly ChIP-seq may have evolved from microarray analysis but it advance real-time PCR and array-based methods. ChIP- required a completely new set of analysis tools to make the seq is a counting assay that uses only short reads to align most of the platform. ChIP-seq analysis begins with mapping to the genome, but requires millions of them to provide of trimmed sequence reads to a reference genome. Next, meaningful data. Fortunately the Solexa 1G NGS gave up peaks are found using peak-calling algorithms. To further to 30M 21-35bp reads per run. -

JOURNAL of PROTEOMICS an Official Journal of the European Proteomics Association (Eupa)
JOURNAL OF PROTEOMICS An official journal of the European Proteomics Association (EuPA) AUTHOR INFORMATION PACK TABLE OF CONTENTS XXX . • Description p.1 • Audience p.1 • Impact Factor p.1 • Abstracting and Indexing p.2 • Editorial Board p.2 • Guide for Authors p.5 ISSN: 1874-3919 DESCRIPTION . Journal of Proteomics is aimed at protein scientists and analytical chemists in the field of proteomics, biomarker discovery, protein analytics, plant proteomics, microbial and animal proteomics, human studies, tissue imaging by mass spectrometry, non-conventional and non-model organism proteomics, and protein bioinformatics. The journal welcomes papers in new and upcoming areas such as metabolomics, genomics, systems biology, toxicogenomics, pharmacoproteomics. Journal of Proteomics unifies both fundamental scientists and clinicians, and includes translational research. Suggestions for reviews, webinars and thematic issues are welcome. All manuscripts are strictly peer reviewed and conform the highest ethical standards. Journal of Proteomics is an official journal of the European Proteomics Association (EuPA) and also publishes official EuPA reports and participates in the International Proteomics Tutorial Programme with HUPO and other partners. Benefits to authors We also provide many author benefits, such as free PDFs, a liberal copyright policy, special discounts on Elsevier publications and much more. Please click here for more information on our author services. Please see our Guide for Authors for information on article submission. If you require any further information or help, please visit our Support Center Should you have an idea for a thematic issue, please complete the thematic issue proposal form and send it to the Editorial Office (Ms. Carly Middendorp, [email protected]).