Transcriptome Analysis Using Next-Generation Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
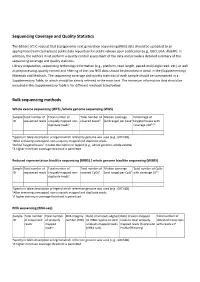
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Microbes and Metagenomics in Human Health an Overview of Recent Publications Featuring Illumina® Technology TABLE of CONTENTS
Microbes and Metagenomics in Human Health An overview of recent publications featuring Illumina® technology TABLE OF CONTENTS 4 Introduction 5 Human Microbiome Gut Microbiome Gut Microbiome and Disease Inflammatory Bowel Disease (IBD) Metabolic Diseases: Diabetes and Obesity Obesity Oral Microbiome Other Human Biomes 25 Viromes and Human Health Viral Populations Viral Zoonotic Reservoirs DNA Viruses RNA Viruses Human Viral Pathogens Phages Virus Vaccine Development 44 Microbial Pathogenesis Important Microorganisms in Human Health Antimicrobial Resistance Bacterial Vaccines 54 Microbial Populations Amplicon Sequencing 16S: Ribosomal RNA Metagenome Sequencing: Whole-Genome Shotgun Metagenomics Eukaryotes Single-Cell Sequencing (SCS) Plasmidome Transcriptome Sequencing 63 Glossary of Terms 64 Bibliography This document highlights recent publications that demonstrate the use of Illumina technologies in immunology research. To learn more about the platforms and assays cited, visit www.illumina.com. An overview of recent publications featuring Illumina technology 3 INTRODUCTION The study of microbes in human health traditionally focused on identifying and 1. Roca I., Akova M., Baquero F., Carlet J., treating pathogens in patients, usually with antibiotics. The rise of antibiotic Cavaleri M., et al. (2015) The global threat of resistance and an increasingly dense—and mobile—global population is forcing a antimicrobial resistance: science for interven- tion. New Microbes New Infect 6: 22-29 1, 2, 3 change in that paradigm. Improvements in high-throughput sequencing, also 2. Shallcross L. J., Howard S. J., Fowler T. and called next-generation sequencing (NGS), allow a holistic approach to managing Davies S. C. (2015) Tackling the threat of anti- microbial resistance: from policy to sustainable microbes in human health. -

Bioinformatics Tools for RNA-Seq Gene and Isoform Quantification
on: Sequ ati en er c n in e g G & t x A Journal of e p Zhang, et al., Next Generat Sequenc & Applic p N l f i c o 2016, 3:3 a l t a i o n r ISSN: 2469-9853n u s DOI: 10.4172/2469-9853.1000140 o Next Generation Sequencing & Applications J Review Article Open Access Bioinformatics Tools for RNA-seq Gene and Isoform Quantification Chi Zhang1, Baohong Zhang1, Michael S Vincent2 and Shanrong Zhao1* 1Early Clinical Development, Pfizer Worldwide R&D, Cambridge, MA, USA 2Inflammation and Immunology RU, Pfizer Worldwide R&D, Cambridge, MA, USA *Corresponding author: Shanrong Zhao, Early Clinical Development, Pfizer Worldwide R&D, Cambridge, MA, 02139, USA, Tel: + 1-212-733-2323; E-mail: [email protected] Rec date: Oct 27, 2016; Acc date: Dec 15, 2016; Pub date: Dec 17, 2016 Copyright: © 2016 Zhang C, et al. This is an open-access article distributed under the terms of the creative commons attribution license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract In recent years, RNA-seq has emerged as a powerful technology in estimation of gene or transcript expression. ‘Union-exon’ and transcript based approaches are widely used in gene quantification. The ‘Union-exon’ based approach is simple, but it does not distinguish between isoforms when multiple alternatively spliced transcripts are expressed from the same gene. Because a gene is expressed in one or more transcript isoforms, the transcript based approach is more biologically meaningful than the ‘union exon’-based approach. -

The Chromosome-Centric Human Proteome Project for Cataloging Proteins Encoded in the Genome
CORRESPONDENCE The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome To the Editor: utility for biological and disease studies. Table 1 Features of salient genes on The Chromosome-Centric Human With development of new tools for in- chromosomes 13 and 17 Proteome Project (C-HPP) aims to define depth characterization of the transcriptome Genea AST nsSNPs the full set of proteins encoded in each and proteome, the HPP is well positioned Chromosome 13 chromosome through development of a to have a strategic role in addressing the BRCA2 3 54 standardized approach for analyzing the complexity of human phenotypes. With this RB1 2 3 massive proteomic data sets currently being in mind, the HUPO has organized national IRS2 1 3 generated from dedicated efforts of national chromosome teams that will collaborate and international teams. The initial goal with well-established laboratories building Chromosome 17 of the C-HPP is to identify at least one complementary proteotypic peptides, BRCA1 24 24 representative protein encoded by each of antibodies and informatics resources. ERBB2 6 13 the approximately 20,300 human genes1,2. An important C-HPP goal is to encourage TP53 14 5 aEnsembl protein and AST information can be found at The proteins will be characterized for tissue capture and open sharing of proteomic http://www.ensembl.org/Homo_sapiens/. localization and major isoforms, including data sets from diverse samples to enhance AST, alternative splicing transcript; nsSNP, nonsyno- mous single-nucleotide polyphorphism assembled from post-translational modifications (PTMs), a gene- and chromosome-centric display data from the 1000 Genomes Projects. -

Downloaded As a CSV Dump file
cells Article Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis Nicolas Buisine 1,† , Alexis Grimaldi 1,†, Vincent Jonchere 1,† , Muriel Rigolet 1, Corinne Blugeon 2 , Juliette Hamroune 2 and Laurent Marc Sachs 1,* 1 UMR7221 Molecular Physiology and Adaption, CNRS, Museum National d’Histoire Naturelle, 57 Rue Cuvier, CEDEX 05, 75231 Paris, France; [email protected] (N.B.); [email protected] (A.G.); [email protected] (V.J.); [email protected] (M.R.) 2 Genomics Core Facility, Département de Biologie, Institut de Biologie de l’ENS (IBENS), École Normale Supérieure, CNRS, INSERM, Université PSL, 75005 Paris, France; [email protected] (C.B.); [email protected] (J.H.) * Correspondence: [email protected] † Co-first authors, alphabetic order. Abstract: Background: Most work in endocrinology focus on the action of a single hormone, and very little on the cross-talks between two hormones. Here we characterize the nature of interactions between thyroid hormone and glucocorticoid signaling during Xenopus tropicalis metamorphosis. Methods: We used functional genomics to derive genome wide profiles of methylated DNA and measured changes of gene expression after hormonal treatments of a highly responsive tissue, tailfin. Clustering classified the data into four types of biological responses, and biological networks were Citation: Buisine, N.; Grimaldi, A.; modeled by system biology. Results: We found that gene expression is mostly regulated by either Jonchere, V.; Rigolet, M.; Blugeon, C.; T or CORT, or their additive effect when they both regulate the same genes. A small but non- Hamroune, J.; Sachs, L.M. -

Glossary of Terms
GLOSSARY OF TERMS Table of Contents A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z A Amino acids: any of a class of 20 molecules that are combined to form proteins in living things. The sequence of amino acids in a protein and hence protein function are determined by the genetic code. From http://www.geneticalliance.org.uk/glossary.htm#C • The building blocks of proteins, there are 20 different amino acids. From https://www.yourgenome.org/glossary/amino-acid Antisense: Antisense nucleotides are strings of RNA or DNA that are complementary to "sense" strands of nucleotides. They bind to and inactivate these sense strands. They have been used in research, and may become useful for therapy of certain diseases (See Gene silencing). From http://www.encyclopedia.com/topic/Antisense_DNA.aspx. Antisense and RNA interference are referred as gene knockdown technologies: the transcription of the gene is unaffected; however, gene expression, i.e. protein synthesis (translation), is lost because messenger RNA molecules become unstable or inaccessible. Furthermore, RNA interference is based on naturally occurring phenomenon known as Post-Transcriptional Gene Silencing. From http://www.ncbi.nlm.nih.gov/probe/docs/applsilencing/ B Biobank: A biobank is a large, organised collection of samples, usually human, used for research. Biobanks catalogue and store samples using genetic, clinical, and other characteristics such as age, gender, blood type, and ethnicity. Some samples are also categorised according to environmental factors, such as whether the donor had been exposed to some substance that can affect health. -

Detection and Characterization of Low and High Genome Coverage Regions Using an Efficient Running Median and a Double Threshold Approach
bioRxiv preprint doi: https://doi.org/10.1101/092478; this version posted December 8, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC 4.0 International license. Bioinformatics doi.10.1093/bioinformatics/xxxxxx Advance Access Publication Date: 2 April 2015 Applications Note Genome analysis Detection and characterization of low and high genome coverage regions using an efficient running median and a double threshold approach. Dimitri Desvillechabrol 1;∗, Christiane Bouchier 2, Sean Kennedy 1, and Thomas Cokelaer 3∗ 1Institut Pasteur – Pole Biomics 2Institut Pasteur – Genomic Platform – Pole Biomics 3Institut Pasteur – Bioinformatics and Biostatistics Hub – C3BI, USR 3756 IP CNRS – Paris, France ∗To whom correspondence should be addressed. Associate Editor: XXXXXXX Received on XXXXX; revised on XXXXX; accepted on XXXXX Abstract Motivation: Next Generation Sequencing (NGS) provides researchers with powerful tools to investigate both prokaryotic and eukaryotic genetics. An accurate assessment of reads mapped to a specific genome consists of inspecting the genome coverage as number of reads mapped to a specific genome location. Most current methods use the average of the genome coverage (sequencing depth) to summarize the overall coverage. This metric quickly assess the sequencing quality but ignores valuable biological information like the presence of repetitive regions or deleted genes. The detection of such information may be challenging due to a wide spectrum of heterogeneous coverage regions, a mixture of underlying models or the presence of a non-constant trend along the genome. -

A Clinically Validated Human Capillary Blood Transcriptome Test for Global Systems Biology Studies
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110080; this version posted May 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A clinically validated human capillary blood transcriptome test for global systems biology studies Ryan Toma1, Ben Pelle2, Nathan Duval1, Matthew M Parks1, Vishakh Gopu1, Hal Tily1, Andrew Hatch1, Ally Perlina1, Guruduth Banavar1, and Momchilo Vuyisich1* 1 Viome, Inc., Bellevue, WA, USA, viome.com, 2 The University of Washington, Seattle, USA *Corresponding author: Momchilo Vuyisich, [email protected] Abstract should be integrated into longitudinal, population- scale, systems biology studies. Chronic diseases are the leading cause of morbidity and mortality globally. Yet, the majority of Introduction them have unknown etiologies, and genetic Quantitative gene expression analysis contribution is weak. In addition, many of the chronic provides a global snapshot of tissue function. The diseases go through the cycles of relapse and human transcriptome varies with tissue type, remission, during which the genomic DNA does not developmental stage, environmental stimuli, and change. This strongly suggests that human gene health/disease state (Frith et al., 2005; Lin et al., 2019; expression is the main driver of chronic disease onset Marioni et al., 2008; Mortazavi et al., 2008; Velculescu and relapses. To identify the etiology of chronic et al., 1999). Changes in the expression patterns can diseases and develop more effective preventative provide insights into the molecular mechanisms of measures, a comprehensive gene expression analysis disease onset and progression. In the era of precision of the human body is needed. -

Bioinformatic Pipelines for Whole Transcriptome Sequencing Data Exploitation in Leukemia Patients with Complex Structural Variants
Bioinformatic pipelines for whole transcriptome sequencing data exploitation in leukemia patients with complex structural variants Jakub Hynst1,2, Karla Plevova1,2,3, Lenka Radova1, Vojtech Bystry1, Karol Pal1,2 and Sarka Pospisilova1,2,3 1 Central European Institute of Technology, Masaryk University, Brno, Czech Republic 2 Department of Internal Medicine—Hematology and Oncology, Faculty of Medicine, Masaryk University, Brno, Czech Republic 3 Department of Internal Medicine—Hematology and Oncology, University Hospital Brno, Brno, Czech Republic ABSTRACT Background. Extensive genome rearrangements, known as chromothripsis, have been recently identified in several cancer types. Chromothripsis leads to complex structural variants (cSVs) causing aberrant gene expression and the formation of de novo fusion genes, which can trigger cancer development, or worsen its clinical course. The functional impact of cSVs can be studied at the RNA level using whole transcriptome sequencing (total RNA-Seq). It represents a powerful tool for discovering, profiling, and quantifying changes of gene expression in the overall genomic context. However, bioinformatic analysis of transcriptomic data, especially in cases with cSVs, is a complex and challenging task, and the development of proper bioinformatic tools for transcriptome studies is necessary. Methods. We designed a bioinformatic workflow for the analysis of total RNA-Seq data Submitted 30 December 2018 consisting of two separate parts (pipelines): The first pipeline incorporates a statistical Accepted 6 May 2019 solution for differential gene expression analysis in a biologically heterogeneous sample Published 12 June 2019 set. We utilized results from transcriptomic arrays which were carried out in parallel to Corresponding authors increase the precision of the analysis. The second pipeline is used for the identification Karla Plevova, of de novo fusion genes. -

2018 Metrics from the HUPO Human Proteome Project † ● ‡ § ∥ Gilbert S
Perspective Cite This: J. Proteome Res. 2018, 17, 4031−4041 pubs.acs.org/jpr Progress on Identifying and Characterizing the Human Proteome: 2018 Metrics from the HUPO Human Proteome Project † ● ‡ § ∥ Gilbert S. Omenn,*, , Lydie Lane, Christopher M. Overall, Fernando J. Corrales, ⊥ # ¶ ∇ ○ Jochen M. Schwenk, Young-Ki Paik, Jennifer E. Van Eyk, Siqi Liu, Michael Snyder, ⊗ ● Mark S. Baker, and Eric W. Deutsch † Department of Computational Medicine and Bioinformatics, University of Michigan, 100 Washtenaw Avenue, Ann Arbor, Michigan 48109-2218, United States ‡ CALIPHO Group, SIB Swiss Institute of Bioinformatics and Department of Microbiology and Molecular Medicine, Faculty of Medicine, University of Geneva, CMU, Michel-Servet 1, 1211 Geneva 4, Switzerland § Life Sciences Institute, Faculty of Dentistry, University of British Columbia, 2350 Health Sciences Mall, Room 4.401, Vancouver, BC V6T 1Z3, Canada ∥ Centro Nacional de Biotecnologia (CSIC), Darwin 3, 28049 Madrid, Spain ⊥ Science for Life Laboratory, KTH Royal Institute of Technology, Tomtebodavagen̈ 23A, 17165 Solna, Sweden # Yonsei Proteome Research Center, Yonsei University, Room 425, Building #114, 50 Yonsei-ro, Seodaemoon-ku, Seoul 120-749, Korea ¶ Advanced Clinical BioSystems Research Institute, Cedars Sinai Precision Biomarker Laboratories, Barbra Streisand Women’s Heart Center, Cedars-Sinai Medical Center, Los Angeles, California 90048, United States ∇ Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, Texas 75390-9148, United States ○ Department -

An Introduction to Next-Generation Sequencing Technology
An introduction to Next-Generation Sequencing Technology www.illumina.com/technology/next-generation-sequencing.html For Research Use Only. Not for use in diagnostic procedures. Table of Contents Table of Contents 2 I. Welcome to Next-Generation Sequencing 3 a. The Evolution of Genomic Science 3 b. The Basics of NGS Chemistry 4 c. Advances in Sequencing Technology 5 Paired-End Sequencing 5 Tunable Coverage and Unlimited Dynamic Range 6 Advances in Library Preparation 6 Multiplexing 7 Flexible, Scalable Instrumentation 7 II. NGS Methods 8 a. Genomics 8 Whole-Genome Sequencing 8 Exome Sequencing 8 De novo Sequencing 9 Targeted Sequencing 9 b. Transcriptomics 11 Total RNA and mRNA Sequencing 11 Targeted RNA Sequencing 11 Small RNA and Noncoding RNASequencing 11 c. Epigenomics 12 Methylation Sequencing 12 ChIP Sequencing 12 Ribosome Profiling 12 III. Illumina DNA-to-Data NGS Solutions 13 a. The Illumina NGS Workflow 13 b. Integrated Data Analysis 13 IV. Glossary 14 V. References 15 For Research Use Only. Not for use in diagnostic procedures. I. Welcome to Next-Generation Sequencing a. The Evolution of Genomic Science DNA sequencing has come a long way since the days of two-dimensional chromatography in the 1970s. With the advent of the Sanger chain termination method1 in 1977, scientists gained the ability to sequence DNA in a reliable, reproducible manner. A decade later, Applied Biosystems introduced the first automated, capillary electrophoresis (CE)-based sequencing instruments,the AB370 in 1987 and the AB3730xl in 1998,instruments that became the primary workhorses for the NIH-led and Celera-led Human Genome Projects.2 While these “first-generation” instruments were considered high throughput for their time, the Genome Analyzer emerged in 2005 and took sequencing runs from 84 kilobase (kb) per run to 1 gigabase (Gb) per run.3 The short read, massively parallel sequencing technique was a fundamentally different approach that revolutionized sequencing capabilities and launched the “next generation” in genomic science. -

Improving Genome Assembly and Annotation Using Transcriptome Data Simo V
Zhang et al. GigaScience (2016) 5:31 DOI 10.1186/s13742-016-0136-3 TECHNICAL NOTE Open Access AGOUTI: improving genome assembly and annotation using transcriptome data Simo V. Zhang1*, Luting Zhuo1 and Matthew W. Hahn1,2 Abstract Background: Genomes sequenced using short-read, next-generation sequencing technologies can have many errors and may be fragmented into thousands of small contigs. These incomplete and fragmented assemblies lead to errors in gene identification, such that single genes spread across multiple contigs are annotated as separate gene models. Such biases can confound inferences about the number and identity of genes within species, as well as gene gain and loss between species. Results: We present AGOUTI (Annotated Genome Optimization Using Transcriptome Information), a tool that uses RNA sequencing data to simultaneously combine contigs into scaffolds and fragmented gene models into single models. We show that AGOUTI improves both the contiguity of genome assemblies and the accuracy of gene annotation, providing updated versions of each as output. Running AGOUTI on both simulated and real datasets, we show that it is highly accurate and that it achieves greater accuracy and contiguity when compared with other existing methods. Conclusion: AGOUTI is a powerful and effective scaffolder and, unlike most scaffolders, is expected to be more effective in larger genomes because of the commensurate increase in intron length. AGOUTI is able to scaffold thousands of contigs while simultaneously reducing the number of gene models by hundreds or thousands. The software is available free of charge under the MIT license. Keywords: Genome assembly, Scaffolding, Genome annotation, RNA sequencing, RNA-seq Background equivalent to intron length.