An Introduction to Next-Generation Sequencing Technology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of the Impact of Sequencing Errors on Blast Using Fault Injection
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Illinois Digital Environment for Access to Learning and Scholarship Repository ANALYSIS OF THE IMPACT OF SEQUENCING ERRORS ON BLAST USING FAULT INJECTION BY SO YOUN LEE THESIS Submitted in partial fulfillment of the requirements for the degree of Master of Science in Electrical and Computer Engineering in the Graduate College of the University of Illinois at Urbana-Champaign, 2013 Urbana, Illinois Adviser: Professor Ravishankar K. Iyer ABSTRACT This thesis investigates the impact of sequencing errors in post-sequence computational analyses, including local alignment search and multiple sequence alignment. While the error rates of sequencing technology are commonly reported, the significance of these numbers cannot be fully grasped without putting them in the perspective of their impact on the downstream analyses that are used for biological research, forensics, diagnosis of diseases, etc. I approached the quantification of the impact using fault injection. Faults were injected in the input sequence data, and the analyses were run. Change in the output of the analyses was interpreted as the impact of faults, or errors. Three commonly used algorithms were used: BLAST, SSEARCH, and ProbCons. The main contributions of this work are the application of fault injection to the reliability analysis in bioinformatics and the quantitative demonstration that a small error rate in the sequence data can alter the output of the analysis in a significant way. BLAST and SSEARCH are both local alignment search tools, but BLAST is a heuristic implementation, while SSEARCH is based on the optimal Smith-Waterman algorithm. -

Advancing Solutions to the Carbohydrate Sequencing Challenge † † † ‡ § Christopher J
Perspective Cite This: J. Am. Chem. Soc. 2019, 141, 14463−14479 pubs.acs.org/JACS Advancing Solutions to the Carbohydrate Sequencing Challenge † † † ‡ § Christopher J. Gray, Lukasz G. Migas, Perdita E. Barran, Kevin Pagel, Peter H. Seeberger, ∥ ⊥ # ⊗ ∇ Claire E. Eyers, Geert-Jan Boons, Nicola L. B. Pohl, Isabelle Compagnon, , × † Göran Widmalm, and Sabine L. Flitsch*, † School of Chemistry & Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester M1 7DN, U.K. ‡ Institute for Chemistry and Biochemistry, Freie Universitaẗ Berlin, Takustraße 3, 14195 Berlin, Germany § Biomolecular Systems Department, Max Planck Institute for Colloids and Interfaces, Am Muehlenberg 1, 14476 Potsdam, Germany ∥ Department of Biochemistry, Institute of Integrative Biology, University of Liverpool, Crown Street, Liverpool L69 7ZB, U.K. ⊥ Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia 30602, United States # Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States ⊗ Institut Lumierè Matiere,̀ UMR5306 UniversitéLyon 1-CNRS, Universitéde Lyon, 69622 Villeurbanne Cedex, France ∇ Institut Universitaire de France IUF, 103 Blvd St Michel, 75005 Paris, France × Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, S-106 91 Stockholm, Sweden *S Supporting Information established connection between their structure and their ABSTRACT: Carbohydrates possess a variety of distinct function, full characterization of unknown carbohydrates features with -
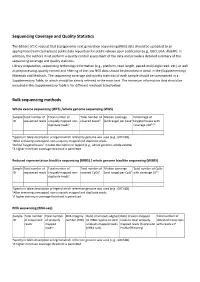
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Grant Application Resources for Visium Spatial Gene Expression
10x Genomics | Visium | Spatial Gene Expression Grant Application Resources Grant application resources for Visium Spatial Gene Expression Summary statement Visium Spatial Gene Expression from 10x Genomics is a novel assay that combines histology with spatially resolved whole transcriptome gene expression profiling to localize and quantify gene expression in the tissue context. It is based on the Spatial Transcriptomics methodology (1). The assay has been well adopted, being utilized in almost 40 peer-reviewed publications and over 50 pre-prints. Currently, Visium Spatial Gene Expression is compatible with fresh frozen tissue sections from any species. This assay utilizes poly(A) capture and novel spatial barcoding technology for library preparation. 10x Genomics also offers a Visium Spatial Gene Expression assay compatible with human and mouse formalin-fixed paraffin-embedded (FFPE) tissue sections. This assay utilizes RNA-templated ligation of pairs of gene target probes for highly specific and sensitive detection of the whole transcriptome. Both assays leverage the same suite of analysis tools and pipelines (e.g., Space Ranger, Loupe Browser) to process and visualize Visium Spatial data. Additionally, researchers have access to 10x Genomics technical experts who can provide support through scientific and technical consultations, workflow optimization, and methodology troubleshooting. Overview The ability to detect and count transcripts by sequencing (RNA-seq) has led to significant advances in our understanding of biology (2), as well as the development of clinical applications. However, traditional RNA-seq suffers from the loss of spatial information. Researchers typically extract RNA from tissue and sequence it in bulk. Data regarding the type of cells expressing a given transcript, the location of these cells within the tissue, and co-expression of transcripts in the tissue geography are lost by this bulk preparation of RNA. -

Whole Genome Sequencing Data of Multiple Individuals of Pakistani
www.nature.com/scientificdata oPeN Whole genome sequencing data DAtA DeScriptor of multiple individuals of Pakistani descent Shahid Y. Khan1, Muhammad Ali1, Mei-Chong W. Lee2, Zhiwei Ma3, Pooja Biswas4, Asma A. Khan5, Muhammad Asif Naeem5, Saima Riazuddin 6, Sheikh Riazuddin5,7,8, Radha Ayyagari4, J. Fielding Hejtmancik 3 & S. Amer Riazuddin1 ✉ Here we report whole genome sequencing of four individuals (H3, H4, H5, and H6) from a family of Pakistani descent. Whole genome sequencing yielded 1084.92, 894.73, 1068.62, and 1005.77 million mapped reads corresponding to 162.73, 134.21, 160.29, and 150.86 Gb sequence data and 52.49x, 43.29x, 51.70x, and 48.66x average coverage for H3, H4, H5, and H6, respectively. We identifed 3,529,659, 3,478,495, 3,407,895, and 3,426,862 variants in the genomes of H3, H4, H5, and H6, respectively, including 1,668,024 variants common in the four genomes. Further, we identifed 42,422, 39,824, 28,599, and 35,206 novel variants in the genomes of H3, H4, H5, and H6, respectively. A major fraction of the variants identifed in the four genomes reside within the intergenic regions of the genome. Single nucleotide polymorphism (SNP) genotype based comparative analysis with ethnic populations of 1000 Genomes database linked the ancestry of all four genomes with the South Asian populations, which was further supported by mitochondria based haplogroup analysis. In conclusion, we report whole genome sequencing of four individuals of Pakistani descent. Background & Summary The completion of Human Genome Project ignited several large scale efforts to characterize variations in the human genome, which led to a comprehensive catalog of the common variants including single-nucleotide polymorphisms (SNPs) and insertions/deletions (indels), across the entire human genome1,2. -

"Phylogenetic Analysis of Protein Sequence Data Using The
Phylogenetic Analysis of Protein Sequence UNIT 19.11 Data Using the Randomized Axelerated Maximum Likelihood (RAXML) Program Antonis Rokas1 1Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee ABSTRACT Phylogenetic analysis is the study of evolutionary relationships among molecules, phenotypes, and organisms. In the context of protein sequence data, phylogenetic analysis is one of the cornerstones of comparative sequence analysis and has many applications in the study of protein evolution and function. This unit provides a brief review of the principles of phylogenetic analysis and describes several different standard phylogenetic analyses of protein sequence data using the RAXML (Randomized Axelerated Maximum Likelihood) Program. Curr. Protoc. Mol. Biol. 96:19.11.1-19.11.14. C 2011 by John Wiley & Sons, Inc. Keywords: molecular evolution r bootstrap r multiple sequence alignment r amino acid substitution matrix r evolutionary relationship r systematics INTRODUCTION the baboon-colobus monkey lineage almost Phylogenetic analysis is a standard and es- 25 million years ago, whereas baboons and sential tool in any molecular biologist’s bioin- colobus monkeys diverged less than 15 mil- formatics toolkit that, in the context of pro- lion years ago (Sterner et al., 2006). Clearly, tein sequence analysis, enables us to study degree of sequence similarity does not equate the evolutionary history and change of pro- with degree of evolutionary relationship. teins and their function. Such analysis is es- A typical phylogenetic analysis of protein sential to understanding major evolutionary sequence data involves five distinct steps: (a) questions, such as the origins and history of data collection, (b) inference of homology, (c) macromolecules, developmental mechanisms, sequence alignment, (d) alignment trimming, phenotypes, and life itself. -

EMBL-EBI Powerpoint Presentation
Processing data from high-throughput sequencing experiments Simon Anders Use-cases for HTS · de-novo sequencing and assembly of small genomes · transcriptome analysis (RNA-Seq, sRNA-Seq, ...) • identifying transcripted regions • expression profiling · Resequencing to find genetic polymorphisms: • SNPs, micro-indels • CNVs · ChIP-Seq, nucleosome positions, etc. · DNA methylation studies (after bisulfite treatment) · environmental sampling (metagenomics) · reading bar codes Use cases for HTS: Bioinformatics challenges Established procedures may not be suitable. New algorithms are required for · assembly · alignment · statistical tests (counting statistics) · visualization · segmentation · ... Where does Bioconductor come in? Several steps: · Processing of the images and determining of the read sequencest • typically done by core facility with software from the manufacturer of the sequencing machine · Aligning the reads to a reference genome (or assembling the reads into a new genome) • Done with community-developed stand-alone tools. · Downstream statistical analyis. • Write your own scripts with the help of Bioconductor infrastructure. Solexa standard workflow SolexaPipeline · "Firecrest": Identifying clusters ⇨ typically 15..20 mio good clusters per lane · "Bustard": Base calling ⇨ sequence for each cluster, with Phred-like scores · "Eland": Aligning to reference Firecrest output Large tab-separated text files with one row per identified cluster, specifying · lane index and tile index · x and y coordinates of cluster on tile · for each -

A SARS-Cov-2 Sequence Submission Tool for the European Nucleotide
Databases and ontologies Downloaded from https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab421/6294398 by guest on 25 June 2021 A SARS-CoV-2 sequence submission tool for the European Nucleotide Archive Miguel Roncoroni 1,2,∗, Bert Droesbeke 1,2, Ignacio Eguinoa 1,2, Kim De Ruyck 1,2, Flora D’Anna 1,2, Dilmurat Yusuf 3, Björn Grüning 3, Rolf Backofen 3 and Frederik Coppens 1,2 1Department of Plant Biotechnology and Bioinformatics, Ghent University, 9052 Ghent, Belgium, 1VIB Center for Plant Systems Biology, 9052 Ghent, Belgium and 2University of Freiburg, Department of Computer Science, Freiburg im Breisgau, Baden-Württemberg, Germany ∗To whom correspondence should be addressed. Associate Editor: XXXXXXX Received on XXXXX; revised on XXXXX; accepted on XXXXX Abstract Summary: Many aspects of the global response to the COVID-19 pandemic are enabled by the fast and open publication of SARS-CoV-2 genetic sequence data. The European Nucleotide Archive (ENA) is the European recommended open repository for genetic sequences. In this work, we present a tool for submitting raw sequencing reads of SARS-CoV-2 to ENA. The tool features a single-step submission process, a graphical user interface, tabular-formatted metadata and the possibility to remove human reads prior to submission. A Galaxy wrap of the tool allows users with little or no bioinformatic knowledge to do bulk sequencing read submissions. The tool is also packed in a Docker container to ease deployment. Availability: CLI ENA upload tool is available at github.com/usegalaxy- eu/ena-upload-cli (DOI 10.5281/zenodo.4537621); Galaxy ENA upload tool at toolshed.g2.bx.psu.edu/view/iuc/ena_upload/382518f24d6d and https://github.com/galaxyproject/tools- iuc/tree/master/tools/ena_upload (development) and; ENA upload Galaxy container at github.com/ELIXIR- Belgium/ena-upload-container (DOI 10.5281/zenodo.4730785) Contact: [email protected] 1 Introduction Nucleotide Archive (ENA). -

MYC-Containing Amplicons in Acute Myeloid Leukemia: Genomic Structures, Evolution, and Transcriptional Consequences
Leukemia (2018) 32:2152–2166 https://doi.org/10.1038/s41375-018-0033-0 ARTICLE Acute myeloid leukemia Corrected: Correction MYC-containing amplicons in acute myeloid leukemia: genomic structures, evolution, and transcriptional consequences 1 1 2 2 1 Alberto L’Abbate ● Doron Tolomeo ● Ingrid Cifola ● Marco Severgnini ● Antonella Turchiano ● 3 3 1 1 1 Bartolomeo Augello ● Gabriella Squeo ● Pietro D’Addabbo ● Debora Traversa ● Giulia Daniele ● 1 1 3 3 4 Angelo Lonoce ● Mariella Pafundi ● Massimo Carella ● Orazio Palumbo ● Anna Dolnik ● 5 5 6 2 7 Dominique Muehlematter ● Jacqueline Schoumans ● Nadine Van Roy ● Gianluca De Bellis ● Giovanni Martinelli ● 3 4 8 1 Giuseppe Merla ● Lars Bullinger ● Claudia Haferlach ● Clelia Tiziana Storlazzi Received: 4 August 2017 / Revised: 27 October 2017 / Accepted: 13 November 2017 / Published online: 22 February 2018 © The Author(s) 2018. This article is published with open access Abstract Double minutes (dmin), homogeneously staining regions, and ring chromosomes are vehicles of gene amplification in cancer. The underlying mechanism leading to their formation as well as their structure and function in acute myeloid leukemia (AML) remain mysterious. We combined a range of high-resolution genomic methods to investigate the architecture and expression pattern of amplicons involving chromosome band 8q24 in 23 cases of AML (AML-amp). This 1234567890();,: revealed that different MYC-dmin architectures can coexist within the same leukemic cell population, indicating a step-wise evolution rather than a single event origin, such as through chromothripsis. This was supported also by the analysis of the chromothripsis criteria, that poorly matched the model in our samples. Furthermore, we found that dmin could evolve toward ring chromosomes stabilized by neocentromeres. -

Genomic Data Standards Resources and Initiatives Cited in the Supplemental Information to the Genomic Data Sharing Policy
Genomic Data Standards Resources and Initiatives Cited in the Supplemental Information to the Genomic Data Sharing Policy IMPORTANT NOTE: The National Institutes of Health makes no endorsement of the non-NIH-funded genomic data standards resources and/or initiatives included in this document. Categories Resources and Initiatives Common Data Element Resource Portal: http://www.nlm.nih.gov/cde/ NIH encourages the use of common data elements (CDEs) in clinical research, patient registries, and other human subject research in order to improve data quality and opportunities for comparison and combination of data from multiple studies and with electronic health records. This portal provides access to NIH-supported CDE initiatives and other tools and resources that can assist investigators developing protocols for data collection. Clinical Genome Resource (ClinGen): http://www.nih.gov/news/health/sep2013/nhgri-25.htm In 2013, the NIH National Human Genome Research Institute (NHGRI) and the Eunice Kennedy Shriver National Institute NIH-Funded of Child Health and Human Development (NICHD) awarded three grants totalling over $25 million to support a consortium of research groups to design and implement a framework for evaluating variants that are relevant to patient care (e.g., play a role in disease). International Collaboration for Clinical Genomics (ICCG): http://www.iccg.org/about-the-iccg/clingen/ ICCG was awarded as a grant under ClinGen. ICCG was charged with developing standard formats for gathering and depositing data in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). It will work with a variety of different stakeholder groups, including clinical laboratories and existing locus-specific databases, to obtain robust data sets on genomic variants and disease associations. -

Comparative Analysis of Pacbio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus Monodon
life Brief Report Comparative Analysis of PacBio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus monodon Zulema Udaondo 1 , Kanchana Sittikankaew 2, Tanaporn Uengwetwanit 2 , Thidathip Wongsurawat 1,3 , Chutima Sonthirod 4, Piroon Jenjaroenpun 1,3 , Wirulda Pootakham 4, Nitsara Karoonuthaisiri 2 and Intawat Nookaew 1,* 1 Department of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA; [email protected] (Z.U.); [email protected] (T.W.); [email protected] (P.J.) 2 National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency, Pathum Thani 12120, Thailand; [email protected] (K.S.); [email protected] (T.U.); [email protected] (N.K.) 3 Division of Bioinformatics and Data Management for Research, Department of Research and Development, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand 4 National Omics Center (NOC), National Science and Technology Development Agency (NSTDA), Pathum Thani 12120, Thailand; [email protected] (C.S.); [email protected] (W.P.) * Correspondence: [email protected]; Tel.: +1-501-686-6025; Fax: +1-501-603-1766 Citation: Udaondo, Z.; Sittikankaew, K.; Uengwetwanit, T.; Wongsurawat, Abstract: With the advantages that long-read sequencing platforms such as Pacific Biosciences T.; Sonthirod, C.; Jenjaroenpun, P.; (Menlo Park, CA, USA) (PacBio) and Oxford Nanopore Technologies (Oxford, UK) (ONT) can offer, Pootakham, W.; Karoonuthaisiri, N.; various research fields such as genomics and transcriptomics can exploit their benefits. Selecting an Nookaew, I. Comparative Analysis of appropriate sequencing platform is undoubtedly crucial for the success of the research outcome, PacBio and Oxford Nanopore thus there is a need to compare these long-read sequencing platforms and evaluate them for specific Sequencing Technologies for research questions. -

Globalfungi, a Global Database of Fungal Occurrences from High
www.nature.com/scientificdata OPEN GlobalFungi, a global database DATA DEscrIPTor of fungal occurrences from high-throughput-sequencing metabarcoding studies Tomáš Větrovský1,6, Daniel Morais1,6, Petr Kohout1,6, Clémentine Lepinay1,6, Camelia Algora1, Sandra Awokunle Hollá1, Barbara Doreen Bahnmann1, Květa Bílohnědá1, Vendula Brabcová1, Federica D’Alò2, Zander Rainier Human1, Mayuko Jomura 3, Miroslav Kolařík1, Jana Kvasničková1, Salvador Lladó1, Rubén López-Mondéjar1, Tijana Martinović1, Tereza Mašínová1, Lenka Meszárošová1, Lenka Michalčíková1, Tereza Michalová1, Sunil Mundra4,5, Diana Navrátilová1, Iñaki Odriozola 1, Sarah Piché-Choquette 1, Martina Štursová1, Karel Švec1, Vojtěch Tláskal 1, Michaela Urbanová1, Lukáš Vlk1, Jana Voříšková1, Lucia Žifčáková1 & Petr Baldrian 1 ✉ Fungi are key players in vital ecosystem services, spanning carbon cycling, decomposition, symbiotic associations with cultivated and wild plants and pathogenicity. The high importance of fungi in ecosystem processes contrasts with the incompleteness of our understanding of the patterns of fungal biogeography and the environmental factors that drive those patterns. To reduce this gap of knowledge, we collected and validated data published on the composition of soil fungal communities in terrestrial environments including soil and plant-associated habitats and made them publicly accessible through a user interface at https://globalfungi.com. The GlobalFungi database contains over 600 million observations of fungal sequences across > 17 000 samples with geographical locations and additional metadata contained in 178 original studies with millions of unique nucleotide sequences (sequence variants) of the fungal internal transcribed spacers (ITS) 1 and 2 representing fungal species and genera. The study represents the most comprehensive atlas of global fungal distribution, and it is framed in such a way that third-party data addition is possible.